一种光学纯汉防己甲素的全合成方法与流程
本发明涉及药物合成技术领域,尤其涉及一种光学纯汉防己甲素的全合成方法。
背景技术:
汉防己甲素(tetrandrine)是从防己科植物粉防己的根中提取的一种生物碱,其结构如下所示,
它有多方面的药理作用,比如解热、镇痛、抗炎、利尿、抗高血压等。诸多文献报道,汉防己甲素对多种肿瘤细胞亦有抑制活性,比如肝癌、肺癌、胃癌、前列腺癌、乳腺癌等。
目前,汉防己甲素一般是通过对汉防己的根进行提取分离来生产汉防己甲素。但是,汉防己根中总生物碱含量为1.5-2.3%,而汉防己甲素的含量约为1%。因此,通过从汉防己根中提取的方法来生产汉防己甲素具有以下缺点:1、原材料汉防己需求量大,生产受限,导致市场供不应求;2、提取工艺复杂,生产效率较低且成本居高不下;3、生产废渣量大,环保压力大。而化学合成法生产药物具有以下优点:1、原料廉价易得,不受限制;2、生产中相关原料、中间体、溶剂等可多次回收套用,经济环保;3、生产量大,可连续生产,节约成本。所以,开发化学合成法生产汉防己甲素具有十分重要的意义。而汉防己甲素的全合成研究对于开发汉防己甲素的化学合成生产工艺具有十分重要的指导作用。
如前所示的结构,天然产物汉防己甲素有两个均为s构型的手性中心。目前,仅有j.chem.soc.1969,1547-1556报道了一种汉防己甲素的全合成方法,该方法路线冗长,操作繁琐,且拆分效率低下,总收率低,仅为20%左右,难以实现工业化生产,且仅为消旋体合成。因此,开发一条可工业化的光学纯汉防己甲素全合成路线,具有重大意义。
本申请的申请人在前期发明了一种高效、经济的消旋汉防己甲素的合成路线,其具体路线为:以95%纯度的5-溴香兰素为起始原料,经甲基化、henry反应和还原,与3-苄氧基-4-甲氧基-苯乙酸缩合,形成的酰胺再经环合、还原、甲基化及脱苄反应,得到了消旋体片段a。以工业品香兰素为起始原料,经苄基保护、henry反应和还原,得到的中间体与对溴苯乙酸缩合后,再经环合、还原、甲基化反应,得到消旋片段b。得到的a和b两个消旋中间体经过第一步ullmann反应进行缩合,再脱去苄基保护基,最后再经ullmann反应完成大环醚键的构建得到消旋的汉防己甲素。
而消旋汉防己甲素的生物活性较低,所以,我们的最终目的是要开发出光学纯汉防己甲素;在消旋汉防己甲素经济高效的合成基础上,开发出光学纯汉防己甲素的合成方法对汉防己甲素的化学合成生产工艺的开发具有重要意义。
技术实现要素:
本发明的目的就在于提供一种光学纯汉防己甲素的全合成方法,以解决上述问题。
为了实现上述目的,本发明采用的技术方案是这样的:一种光学纯汉防己甲素的全合成方法,包括以下步骤:
(1)化合物1与化合物2在催化剂1作用下,碱性、高温条件下经过分子间ullmann反应合成化合物3;
(2)化合物3在酸性条件下脱去羟基保护基后合成化合物4;
(3)化合物4在催化剂2作用下,碱性、高温条件下经过分子内ullmann反应合成化合物5,即光学纯汉防己甲素,其合成路线如下所示:
本发明的化合物1和化合物2,可以是采用化学拆分的方法,将消旋化合物拆分成光学纯化合物,因为其消旋体更易得,具体的拆分方法可以是如下方法:
(1)化合物1的制备
盐的制备:室温下,100ml圆底烧瓶中加入3g化合物1的消旋体和50g乙醇,将0.8gn-ac-l-半胱氨酸溶解在20g乙醇后加入化合物1的消旋体的乙醇溶液中,0℃析晶12h后过滤,滤饼用乙醇洗涤两次后得到白色固体;
精制:50ml圆底烧瓶中加入得到的白色固体与20g80%乙醇,加热至60℃搅洗半小时,趁热过滤,滤饼用80%乙醇洗涤2遍,滤饼经干燥后得到白色固体;
解离:将精制产物溶解于二氯甲烷中,加入10%氢氧化钠溶液5ml,静置分液后水相再用50g二氯甲烷萃取两次;合并二氯甲烷相,用50g水洗两次,无水硫酸钠干燥。过滤,滤液减压浓缩至干,得白色固体1.3g,经过核磁氢谱、碳谱和标准品对照,确认为化合物1,ee值为99.89%;
(2)化合物2的制备
盐的制备:室温下,250ml圆底烧瓶中加入6.4g化合物2的消旋体和90g乙醇,将1.7gn-ac-l-半胱氨酸溶解在50g乙醇后加入化合物2消旋体的乙醇溶液中,0℃析晶24h后过滤,滤饼用乙醇洗涤两次后得到白色固体;
精制:100ml圆底烧瓶中加入得到的白色固体与40g90%乙醇,加热至55℃搅洗半小时,趁热过滤,滤饼用90%乙醇洗涤2遍,滤饼经干燥后得到白色固体;
解离:将精制产物溶解于二氯甲烷中,加入10%氢氧化钠溶液10ml,静置分液后水相再用50g二氯甲烷萃取两次。合并二氯甲烷相,用50g水洗两次,无水硫酸钠干燥。过滤,滤液减压浓缩至干,得白色固体2.7g,经过核磁氢谱、碳谱和标准品对照,确认为化合物2,ee值为99.95%。
当然,本领域技术人员可以理解的,化合物1和2的获得,不仅限于上述方法,上述方法仅是例举而非穷举。
作为优选的技术方案:所述步骤(1)中,催化剂选自氯化亚铜、溴化亚铜、碘化亚铜、铜粉、氧化铜、氧化亚铜、或溴化亚铜二甲硫醚络合物中的一种;碱性试剂选自氢氧化钠、氢氧化钾、碳酸钾、碳酸铯或三乙胺中的一种;溶剂选自二氧六环、吡啶、邻二甲苯、甲苯、dmf或dmso中的一种。
作为进一步优选的技术方案:步骤(1)中,催化剂为溴化亚铜;碱性试剂为碳酸钾;溶剂为甲苯。
作为优选的技术方案:所述步骤(1)中,化合物1与化合物2的摩尔比为1:0.5-1:2,化合物1与碱性试剂的摩尔比为1:1-1:5,催化剂用量为化合物1重量的5%-50%,反应温度为加热至回流,反应时间为24-72小时。
作为优选的技术方案:所述步骤(2)中,酸性试剂选自硫酸、醋酸、三氟醋酸、盐酸或氢溴酸中的一种;溶剂选自甲醇、乙醇或异丙醇中的一种。
作为进一步优选的技术方案:所述步骤(2)中,酸性试剂为硫酸;溶剂为甲醇。
作为优选的技术方案:所述步骤(2)中,化合物3与酸性试剂的摩尔比为1:20-1:200,反应温度为加热至回流,反应时间为4-10小时。
作为优选的技术方案:所述步骤(3)中,催化剂选自氯化亚铜、溴化亚铜、碘化亚铜、铜粉、氧化铜、氧化亚铜、或溴化亚铜二甲硫醚络合物中的一种;碱性试剂选自氢氧化钠、氢氧化钾、碳酸钾、碳酸铯或三乙胺中的一种;溶剂选自二氧六环、吡啶、邻二甲苯、甲苯、dmf或dmso中的一种。
作为进一步优选的技术方案:所述步骤(3)中,催化剂为碘化亚铜;碱性试剂为碳酸铯;溶剂为dmf。
作为优选的技术方案:所述步骤(3)中,化合物4与碱性试剂的摩尔比为1:1-1:5,催化剂用量为化合物4重量的5%-50%,反应温度为加热至回流,反应时间为24-72小时。
通过上述工艺参数的优化,可以使得率和产物纯度达到更优。
与现有技术相比,本发明的优点在于:
1、反应步骤明显缩短
现有技术中,采用的是“线性”的合成策略,从原料出发到合成消旋汉防己甲素一共需要9步,步骤繁琐,时间和物料成本高,
而本发明采用“汇聚”式合成的策略,从化合物1出发到合成光学纯汉防己甲素(化合物5)仅需要3步,大大缩减了反应步骤,节约时间和物料成本;
2、反应收率明显提高
现有技术从原料合成消旋汉防己甲素的收率仅为1.67%,
而本发明因反应步骤的大大缩减,从化合物1合成光学纯汉防己甲素(化合物5)的收率可以高达28.7%-38.9%,收率提高数十倍;
3、反应规模的扩大
现有技术仅为毫克级合成,最终得到消旋体天然产物仅为20mg,
本发明能以克级规模得到目标产物,合成最终光学纯天然产物1.3g-1.5g,更具有工业化潜力,这一方面得益于本发明的“汇聚式”合成的思路,减少了合成步骤,另一方面也得益于本发明的优化的合适的合成工艺参数。
附图说明
图1为实施例1中化合物3的hplc光学纯度图;
图2为实施例1中化合物4的hplc光学纯度图;
图3为天然提取的汉防己甲素的hplc光学纯度图;
图4为实施例1中化合物5的hplc光学纯度图;
图5为实施例1中化合物5的核磁氢谱;
图6为实施例1中化合物5的核磁碳谱。
具体实施方式
下面将结合附图对本发明作进一步说明。
实施例1
(1)化合物3的制备
250ml三口瓶中加入4.2g化合物1、4.6g化合物2、0.36g溴化亚铜、9.8g碳酸钾和84g甲苯,氮气保护下加热至100℃反应48小时,原料反应完全;反应液减压浓缩至干,加入42g水,用10%稀盐酸调节ph=3左右,再用82g二氯甲烷萃取两次;合并二氯甲烷相,用82g水洗两次,无水硫酸钠干燥;过滤,滤液减压浓缩至干,得棕色固体,通过柱层析纯化,得淡黄色固体5.5g,即为化合物3,收率69.7%,ee值为99.2%,hplc检测其光学纯度的色谱条件为:流动相比例:0.1%二乙胺-正己烷:乙醇=80:20;流速:1.0ml/min;柱温:30℃;波长:220nm;进样量:20ul;运行时间:40min;色谱柱:ad-h;进样量:0.3mg/ml,结果见图1;
(2)化合物4的制备
250ml三口瓶中加入4g化合物2、50g硫酸和80g甲醇,氮气保护下加热至回流反应6小时,原料反应完全;反应液减压浓缩至干,加入80g水,缓慢滴加饱和碳酸氢钠溶液调节ph=8左右,用100g二氯甲烷萃取两次。合并二氯甲烷相,80g水洗两次,无水硫酸钠干燥。过滤,滤液减压浓缩至干,得白色固体3.5g,即为化合物4,收率98.7%,ee值为99.3%,hplc检测其光学纯度的色谱条件为:流动相比例:0.1%二乙胺-正己烷:乙醇=75:25;流速:0.8ml/min;柱温:30℃;波长:220nm;进样量:20ul;运行时间:35min;色谱柱:ad-h;进样量:0.3mg/ml,结果见图2;
(3)光学纯汉防己甲素(化合物5)的制备
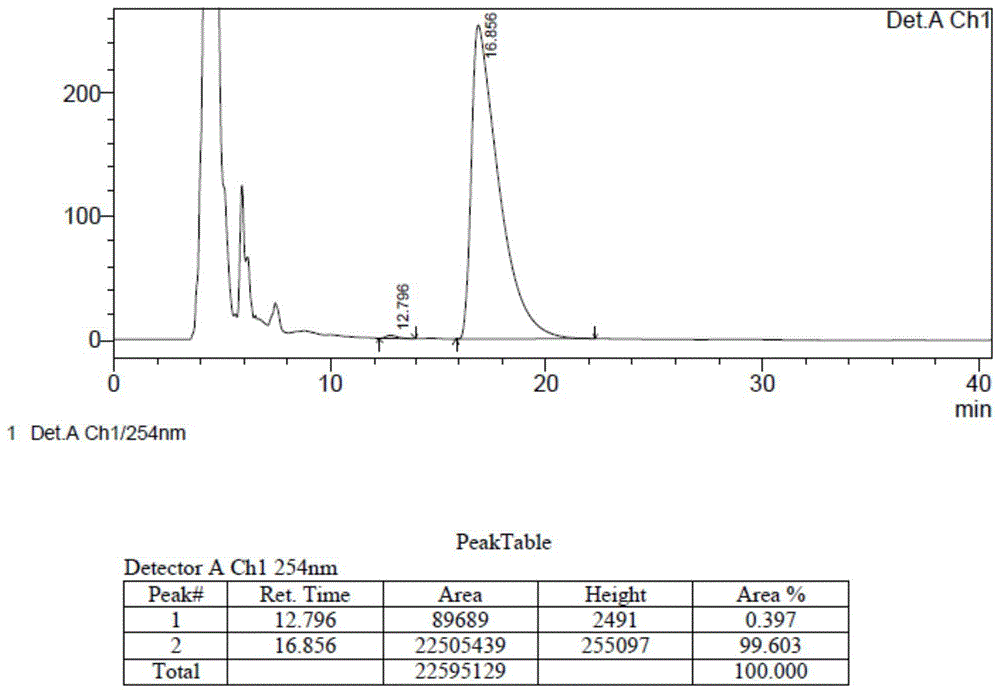
500ml三口瓶中加入3g化合物4、0.6g碘化亚铜、4.2g碳酸铯和150g甲苯,氮气保护下加热至回流反应80小时,原料反应完全。反应液减压浓缩至干,加入50g水,用10%稀盐酸调节ph=3左右,再用100g二氯甲烷萃取两次;合并二氯甲烷相,用100g水洗两次,无水硫酸钠干燥;过滤,滤液减压浓缩至干,得棕色固体,通过柱层析纯化,得类白色固体1.5g,即为光学纯汉防己甲素,收率56.5%,ee值为99.7%,hplc检测其光学纯度的色谱条件为:流动相比例:0.1%二乙胺-正己烷:乙醇=90:10;流速:0.8ml/min;柱温:30℃;波长:220nm;进样量:20ul;运行时间:20min;色谱柱:od-h;进样量:0.3mg/ml;乙醇配样;峰高500-1000,结果见图4;
所得产物的结构经过1h-nmr、13c-nmr测试结果见图5、图6,可见与文献报道一致,并采用标准品(见图3)对照进行内标法hplc检测,证实了其结构为光学纯汉防己甲素。
即本实施例,整个合成过程的收率为:69.7%*98.7%*56.5%=38.9%。
实施例2
(1)化合物3的制备
250ml三口瓶中加入4.8g化合物1、4.0g化合物2、1.8g溴化亚铜二甲硫醚混合物、8.4g碳酸铯和80g邻二甲苯,氮气保护下加热至回流反应48小时,原料反应完全。反应液减压浓缩至干,加入50g水,用10%稀盐酸调节ph=3左右,再用100g二氯甲烷萃取两次。合并二氯甲烷相,用100g水洗两次,无水硫酸钠干燥。过滤,滤液减压浓缩至干,得棕色固体,通过柱层析纯化,得淡黄色固体5.1g,即为化合物3,收率56.9%,ee值为99.3%;
(2)化合物4的制备
250ml三口瓶中加入3.0g化合物3、60g盐酸和60g甲醇,氮气保护下加热至回流反应8小时,原料反应完全。反应液减压浓缩至干,加入60g水,缓慢滴加饱和碳酸氢钠溶液调节ph=8左右,用100g二氯甲烷萃取两次。合并二氯甲烷相,100g水洗两次,无水硫酸钠干燥。过滤,滤液减压浓缩至干,得白色固体2.6g,即为化合物4,收率97.8%,ee值为99.7%。
(3)光学纯汉防己甲素(化合物5)的制备
500ml三口瓶中加入2.8g化合物4、0.56g氧化铜、3.6g碳酸铯和150gdmf,氮气保护下加热至110℃反应60小时,原料反应完全。反应液减压浓缩至干,加入50g水,用10%稀盐酸调节ph=3左右,再用100g二氯甲烷萃取两次。合并二氯甲烷相,用100g水洗两次,无水硫酸钠干燥。过滤,滤液减压浓缩至干,得棕色固体,通过柱层析纯化,得类白色固体1.3g,即为光学纯汉防己甲素,收率52.5%,ee值为99.9%。
即,整个合成过程的收率为:56.9%*97.8%*52.5%=29.2%。
实施例3
(1)化合物3的制备
250ml三口瓶中加入4.8g化合物1、4.0g化合物2、0.3g氯化亚铜、2.0g氢氧化钠和80g甲苯,氮气保护下加热至回流反应60小时,原料反应完全。反应液减压浓缩至干,加入50g水,用10%稀盐酸调节ph=3左右,再用100g二氯甲烷萃取两次。合并二氯甲烷相,用100g水洗两次,无水硫酸钠干燥。过滤,滤液减压浓缩至干,得棕色固体,通过柱层析纯化,得淡黄色固体4.9g,即为化合物3,收率54.1%,ee值为98.6%。
(2)化合物4的制备
250ml三口瓶中加入3.0g化合物3、60g盐酸和60g乙醇,氮气保护下加热至回流反应2小时,原料反应完全。反应液减压浓缩至干,加入60g水,缓慢滴加饱和碳酸氢钠溶液调节ph=8左右,用100g二氯甲烷萃取两次。合并二氯甲烷相,100g水洗两次,无水硫酸钠干燥。过滤,滤液减压浓缩至干,得白色固体2.5g,即为化合物4,收率94.0%,ee值为99.6%。
(3)光学纯汉防己甲素(化合物5)的制备
500ml三口瓶中加入2.8g化合物4、0.45g氯化亚铜、3.6g碳酸钾和150gdmf,氮气保护下加热至125℃反应72小时,原料反应完全。反应液减压浓缩至干,加入100g水,用10%稀盐酸调节ph=3左右,再用100g二氯甲烷萃取两次。合并二氯甲烷相,用100g水洗两次,无水硫酸钠干燥。过滤,滤液减压浓缩至干,得棕色固体,通过柱层析纯化,得类白色固体1.4g,即为光学纯汉防己甲素,收率56.5%,ee值为99.5%;
即,整个合成过程的收率为:54.1%*94.0%*56.5%=28.7%。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!