一种1/10水马来酸氯苯那敏化合物及其药物组合物的制作方法
本发明属于医药
技术领域:
,具体涉及一种1/10水马来酸氯苯那敏化合物及其制备方法和药物组合物。
背景技术:
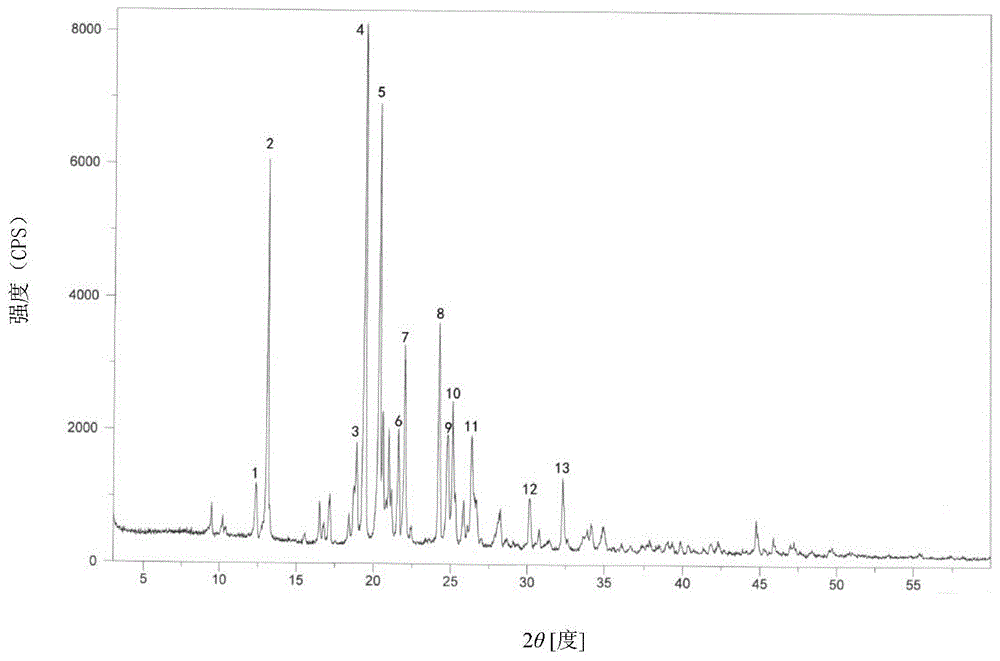
:马来酸氯苯那敏,化学名:2-[对-氯-α-[2-(二甲氨基)乙基]苯基]吡啶马来酸盐,分子式:c16h19cln2·c4h4o4·1/10h2o,分子量为392.67,结构式下:马来酸氯苯那敏作为组织胺h1受体拮抗剂,能对抗过敏反应所致的毛细血管扩张,降低毛细血管的通透性,缓解支气管平滑肌收缩所致的喘息,抗组胺作用较持久,也具有明显的中枢抑制作用,能增加麻醉药、镇痛药、催眠药和局麻药的作用。马来酸氯苯那敏常与复方阿司匹林或其他解热镇痛药配合用于治疗感冒、缓解流泪、打喷嚏、流涕等感冒症状。专利201910709868.1公开了一种马来酸氯苯那敏的合成方法,以对氯苄卤为起始原料,在酸性条件下,在钴催化剂、锰还原剂存在下和2-卤吡啶发生反应,生成2-对氯苄基吡啶,然后在氨基钠的作用下和n,n-二甲基卤乙烷反应,生成氯苯那敏,再和马来酸成盐得到马来酸氯苯那敏。专利201410085339.6公开了一种马来酸氯苯那敏化合物及其药物组合物,所述马来酸氯苯那敏化合物是通过将马来酸氯苯那敏粗品进行精制,得到其新晶体化合物。采用这种新晶型化合物制得布洛伪麻那敏片。其晶体的制备需要多种溶剂,包括毒性较大的氯仿和危险性较高的乙醚,且溶剂用量较大,因此,不利于制剂开发和工业化生产。专利201510968588.4公开了一种新的马来酸氯苯那敏合成方法,以对氯氰苄为起始原料,在催化剂二茂钴的催化下和乙炔气体反应,生成对氯苄基吡啶,对氯苄基吡啶在氨基钠的催化下和n,n-二甲基氯乙烷反应,生成氯苯那敏,然后和马来酸中和反应,生成马来酸氯苯那敏。该路线反应条件苛刻,乙炔气是易燃气体,存在较大安全隐患。专利201911333005.5公开了一种新的马来酸氯苯那敏晶体及其制备方法,将马来酸和氯苯那敏在溶剂中进行反应,制得马来酸氯苯那敏晶体。其在12.4±0.2°、13.1±0.2°、18.8±0.2°、19.3±0.2°、20.2±0.2°、24.9±0.2°、26.5±0.2°有特征峰。目前市场上的马来酸氯苯那敏原料均为无水物,有多种不同的晶型,不同晶型具有不同的物理性质,直接影响制剂的稳定性和生物利用度,而且现有晶型的溶解性和稳定性还有待进一步提高。本发明人以现有技术为基础,经过大量的试验,制得了一种含有1/10水的马来酸氯苯那敏化合物,并通过试验发现1/10水马来酸氯苯那敏化合物具有稳定性好、纯度高、溶解性好、制剂生物利用度高的优点,而且工艺简单,收率高,重复性强,适合于工业化生产。技术实现要素:本发明的目的在于开发和制备一种新的马来酸氯苯那敏化合物产品,克服目前市场上马来酸氯苯那敏原料药的稳定性差、溶解度低、生物利用度低等问题。为解决这些问题,本发明制备出了一种1/10水马来酸氯苯那敏化合物。本发明提供了一种1/10水马来酸氯苯那敏化合物及其制备方法。本发明还提供了一种含有1/10水马来酸氯苯那敏化合物的药物组合物。本发明的技术方案如下:一种1/10水马来酸氯苯那敏化合物,分子式为c16h19cln2·c4h4o4·1/10h2o,分子量为392.67,结构式如下:本发明所述的1/10水马来酸氯苯那敏化合物,其用粉末x射线衍射测定法测定,以2θ±0.2°衍射角表示的x射线粉末衍射图谱如图1所示,在12.4°,13.1°,18.9°,19.4°,20.3°,21.6°,22.0°,24.2°,24.7°,25.1°,26.3°,30.1°,32.3°处显示出特征衍射峰。一种1/10水马来酸氯苯那敏化合物的制备方法,具体包括如下步骤:(1)起始物料4-(2-吡啶甲基)苯胺加浓盐酸、纯化水,低温下滴加亚硝酸钠溶液,搅拌,再滴加氯化亚铜盐酸溶液,室温反应,加入乙酸乙酯萃取,干燥,得中间体i;(2)将中间体i、甲苯、氨基钠混合搅拌,滴加溴代乙醛缩乙二醇,回流,加纯化水淬灭反应,分液,浓缩,得中间体ii;(3)将中间体ii、甲酸、dmf、纯化水混合反应,加入乙酸乙酯萃取,干燥,浓缩,得中间体iii;(4)将中间体iii、马来酸、异丙醇混合反应,降温,搅拌析晶,干燥,得马来酸氯苯那敏化合物。优选地,其步骤(1)中滴加亚硝酸钠溶液需0~5℃,搅拌20min。优选地,其步骤(3)中混合反应条件是100℃,反应24小时。优选地,其步骤(4)中混合反应条件是80℃,反应2小时。优选地,其步骤(4)中降温至室温,搅拌析晶2小时,50℃烘干。作为本发明一优选实施方案,1/10水马来酸氯苯那敏化合物的制备方法具体如下:(1)起始物料4-(2-吡啶甲基)苯胺加浓盐酸、纯化水,降温至0-5℃,滴加亚硝酸钠溶液,滴加完毕,在0-5℃搅拌20min,再滴加氯化亚铜盐酸溶液,室温反应4小时,加入乙酸乙酯萃取,有机相水洗一次,无水硫酸钠干燥,旋干,得中间体i;(2)将中间体i、甲苯、氨基钠混合搅拌20min,滴加溴代乙醛缩乙二醇,滴加完毕,升温回流8小时,降至室温,加纯化水淬灭反应,分液,有机相水洗一次,浓缩,旋干,得中间体ii;(3)将中间体ii、甲酸、dmf、纯化水混合反应,升温至100℃,反应24小时,降至室温,加入乙酸乙酯萃取,分液,有机相水洗一次,无水硫酸钠干燥,浓缩,旋干,得中间体iii;(4)将中间体iii、马来酸、异丙醇混合反应,升温至80℃,反应2小时,降至室温,搅拌析晶2小时,过滤,50℃烘干,得马来酸氯苯那敏化合物。一种药物组合物,其包含本发明所述的1/10水马来酸氯苯那敏化合物和药学上可接受的赋形剂。本发明所述的药物组合物还含有对乙酰氨基酚、咖啡因、人工牛黄等。本发明所述的药物组合物为对乙酰氨基酚、咖啡因、人工牛黄、马来酸氯苯那敏及辅料制成的口服固体制剂。本发明所述的药物组合物口服固体制剂可以为片剂、胶囊剂、颗粒剂等。与现有技术相比,本发明具有如下优点:1、本发明提供的马来酸氯苯那敏化合物纯度高,溶解性好,稳定性好。2、本发明提供的马来酸氯苯那敏化合物制备方法工艺简单,收率高,重复性强,适合于工业化生产。3、本发明提供的含有马来酸氯苯那敏化合物的药物组合物稳定性很好,生物利用度高,大大提高了用药安全性和有效性,降低了不良反应的发生率。附图说明以下,结合附图来详细说明本发明的实施方案,其中:图1为本发明的马来酸氯苯那敏化合物的x射线粉末衍射图。具体实施方式以下实施例是对本发明的进一步说明,但绝不是对本发明范围的限制。下面参照实施例进一步详细阐述本发明,但是本领域技术人员应当理解,本发明并不限于这些实施例以及使用的制备方法。而且,本领域技术人员根据本发明的描述可以对本发明进行等同替换、组合、改良或修饰,但这些都将包括在本发明的范围内。实施例11/10水马来酸氯苯那敏化合物的制备(1)将4-(2-吡啶甲基)苯胺100g,浓盐酸136ml,纯化水200ml,加入反应瓶中,降温至0-5℃,滴加亚硝酸钠溶液(亚硝酸钠38g,纯化水50ml),滴加完毕,在0-5℃搅拌20min,再滴加氯化亚铜盐酸溶液(氯化亚铜135.8g,浓盐酸200ml),室温反应4小时,加入乙酸乙酯700ml萃取,有机相水洗一次,无水硫酸钠干燥,旋干,得中间体i94g;(2)将中间体i90g、甲苯500ml、氨基钠26.6g加入反应瓶中,室温搅拌20min,滴加溴代乙醛缩乙二醇133g,滴加完毕,升温回流8小时,降至室温,加纯化水50ml淬灭反应,分液,有机相水洗一次,浓缩,旋干,得中间体ii110g;(3)将中间体ii110g、甲酸93g、dmf100ml、纯化水200ml加入反应瓶中,升温至100℃,反应24小时,降至室温,每次加入乙酸乙酯500ml,萃取两次,分液,有机相水洗一次,无水硫酸钠干燥,浓缩,旋干,得中间体iii80g;(4)将中间体iii80g、马来酸39.2g、异丙醇400ml加入反应瓶中,升温至80℃,反应2小时,降至室温,搅拌析晶2小时,过滤,50℃烘干,得马来酸氯苯那敏化合物99.6g,收率87.5%。实施例21/10水马来酸氯苯那敏化合物的制备(1)将4-(2-吡啶甲基)苯胺100g,浓盐酸136ml,纯化水200ml,加入反应瓶中,降温至0-5℃,滴加亚硝酸钠溶液(亚硝酸钠38g,纯化水50ml),滴加完毕,在0-5℃搅拌20min,再滴加氯化亚铜盐酸溶液(氯化亚铜135.8g,浓盐酸200ml),室温反应4小时,加入乙酸乙酯700ml萃取,有机相水洗一次,无水硫酸钠干燥,旋干,得中间体i95.1g;(2)将中间体i90g、甲苯500ml、氨基钠26.6g加入反应瓶中,室温搅拌20min,滴加溴代乙醛缩乙二醇133g,滴加完毕,升温回流8小时,降至室温,加纯化水50ml淬灭反应,分液,有机相水洗一次,浓缩,旋干,得中间体ii112.2g;(3)将中间体ii110g、甲酸93g、dmf100ml、纯化水200ml加入反应瓶中,升温至100℃,反应24小时,降至室温,每次加入乙酸乙酯500ml,萃取两次,分液,有机相水洗一次,无水硫酸钠干燥,浓缩,旋干,得中间体iii80.8g;(4)将中间体iii80g、马来酸39.2g、异丙醇400ml加入反应瓶中,升温至80℃,反应2小时,降至室温,搅拌析晶2小时,过滤,50℃烘干,得马来酸氯苯那敏化合物101.1g,收率88.8%。实施例31/10水马来酸氯苯那敏化合物的制备(1)将4-(2-吡啶甲基)苯胺100g,浓盐酸136ml,纯化水200ml,加入反应瓶中,降温至0-5℃,滴加亚硝酸钠溶液(亚硝酸钠38g,纯化水50ml),滴加完毕,在0-5℃搅拌20min,再滴加氯化亚铜盐酸溶液(氯化亚铜135.8g,浓盐酸200ml),室温反应4小时,加入乙酸乙酯700ml萃取,有机相水洗一次,无水硫酸钠干燥,旋干,得中间体i93.3g;(2)将中间体i90g、甲苯500ml、氨基钠26.6g加入反应瓶中,室温搅拌20min,滴加溴代乙醛缩乙二醇133g,滴加完毕,升温回流8小时,降至室温,加纯化水50ml淬灭反应,分液,有机相水洗一次,浓缩,旋干,得中间体ii110.6g;(3)将中间体ii110g、甲酸93g、dmf100ml、纯化水200ml加入反应瓶中,升温至100℃,反应24小时,降至室温,每次加入乙酸乙酯500ml,萃取两次,分液,有机相水洗一次,无水硫酸钠干燥,浓缩,旋干,得中间体iii81.2g;(4)将中间体iii80g、马来酸39.2g、异丙醇400ml加入反应瓶中,升温至80℃,反应2小时,降至室温,搅拌析晶2小时,过滤,50℃烘干,得马来酸氯苯那敏化合物98.5g,收率86.6%。实施例4含有1/10水马来酸氯苯那敏药物组合物的制备处方:制备过程:(1)称取处方量的原辅料,选择性粉碎过筛,使粒度达到要求。(2)称取一定量的蔗糖,加沸水制成蔗糖溶液,分别加入不同颜色的色素,作为粘合剂备用。(3)将对乙酰氨基酚、咖啡因、马来酸氯苯那敏、人工牛黄、蔗糖、玉米淀粉置湿法制粒机内,混合,加入粘合剂,搅拌切碎,制粒。(4)干燥,60-70℃干燥3-4小时。(5)整粒,总混。(6)填充胶囊。(7)包装。实施例5含有1/10水马来酸氯苯那敏药物组合物的制备处方:制备过程:(1)称取处方量的原辅料,选择性粉碎过筛,使粒度达到要求。(2)称取处方量的羟丙甲纤维素,加热水制成5%羟丙甲纤维素溶液,加入酒石酸溶解,作为粘合剂备用。(3)将对乙酰氨基酚、咖啡因、马来酸氯苯那敏、人工牛黄、玉米淀粉置湿法制粒机内,混合,加入粘合剂,搅拌切碎,制粒。(4)干燥。(5)整粒,总混。(6)压片。(7)包装。实施例6含有1/10水马来酸氯苯那敏药物组合物的制备处方:制备过程:(1)称取处方量的原辅料,选择性粉碎过筛,使粒度达到要求。(2)称取处方量的聚维酮k30,加水制成10%聚维酮k30溶液,作为粘合剂备用。(3)将对乙酰氨基酚、马来酸氯苯那敏、人工牛黄、蔗糖置湿法制粒机内,混合,加入粘合剂,搅拌切碎,制粒。(4)干燥。(5)整粒,加入香精,总混。(6)装袋。对比例1马来酸氯苯那敏的制备根据专利201910709868.1公开的一种马来酸氯苯那敏的合成方法。将2-溴吡啶(15.8g,0.1mol)、对氯苄氯(19.3g,0.12mol)、乙腈(80g)和锰粉(12.1g,0.22mol)加入到反应瓶中,再加入三氟乙酸(1ml)活化锰粉。10分钟后加入溴化钴(1.1g,0.005mol),保持温度在30-40℃之间反应12小时。用碳酸钠中和后过滤,滤液减压除去溶剂。残留物用甲苯溶解,5%的edta水溶液洗涤,有机层即2-对氯苄基吡啶甲苯溶液。称取氨基钠(7.8g,0.2mol)加入到50ml甲苯中,再加入2-对氯苄基吡啶甲苯溶液,加热回流,然后慢慢滴加n,n-二甲胺基氯乙烷(21.4g,0.2mol)的甲苯的溶液。回流反应6小时,冷却后过滤得含氯苯那敏盐基的甲苯溶液。加水洗至中性,减压回收甲苯后与顺丁烯二酸成盐,乙醇为溶剂重结晶。得马来酸氯苯那敏白色固体21.0g(纯度98.2%,产率54.4%)。对比例2马来酸氯苯那敏药物组合物的制备采用对比例1制备的马来酸氯苯那敏,处方和制备工艺同实施例4。对比例3马来酸氯苯那敏药物组合物的制备采用对比例1制备的马来酸氯苯那敏,处方和制备工艺同实施例5。对比例4马来酸氯苯那敏药物组合物的制备采用对比例1制备的马来酸氯苯那敏,处方和制备工艺同实施例6。试验例1纯度比较本发明人对本发明实施例1~3和对比例1制备的马来酸氯苯那敏化合物进行了纯度检测,结果如下:实施例实施例1实施例2实施例3对比例1纯度(%)99.999.999.898.2结论:由上表数据可明显看出,本发明实施例1~3制备的1/10水马来酸氯苯那敏化合物纯度明显好于对比例1制备的样品,说明了本发明技术方案对提高马来酸氯苯那敏化合物纯度具有显著效果。试验例2溶解性试验本发明人对本发明实施例1~3和对比例1所制备的马来酸氯苯那敏分别溶于水溶液中,震摇20min,通过检测含量计算实施例1~3、对比例1所制备的马来酸氯苯那敏在水中的溶解度。溶解度检测结果实施例溶解度实施例165mg/ml实施例271mg/ml实施例368mg/ml对比例135mg/ml结论:本发明实施例1~3制备的1/10水马来酸氯苯那敏化合物的溶解性明显好于对比例1的马来酸氯苯那敏化合物。试验例3热稳定性试验本发明人将本发明实施例1~3和对比例1制备的马来酸氯苯那敏化合物置于温度60℃,相对湿度75%条件下30天,分别于0、15、30天进行检测,考察指标为性状、澄清度与颜色、含量和有关物质。结果如下:结论:由上表数据可明显看出,本发明实施例1~3制备的1/10水马来酸氯苯那敏化合物热稳定性明显好于对比例1制备的样品,说明了本发明技术方案对提高马来酸氯苯那敏化合物热稳定性具有显著效果。试验例4稳定性考察本发明人将本发明实施例4~6和对比例2~4制备的含有马来酸氯苯那敏的药物组合物进行加速和长期稳定性考察试验。加速试验考察条件为温度40±2℃,相对湿度75%±5%,放置6个月,分别于0、1、2、3、6月取样;长期试验考察条件为温度25±2℃,相对湿度60%±5%,放置24个月,分别于0、3、6、9、12月取样。考察指标为性状、崩解时限和含量。结果如下:加速试验结果:长期试验结果:结论:由上表数据可明显看出,本发明实施例4~6制备的含有马来酸氯苯那敏的药物组合物稳定性明显好于对比例2~4制备的样品,说明了本发明技术方案对提高含有马来酸氯苯那敏的药物组合物稳定性具有显著效果。试验例51/10水马来酸氯苯那敏化合物的确认为了充分验证本发明马来酸氯苯那敏化合物中的1/10水为结晶水,本发明人通过热重分析法、60℃热稳定性10天、冷冻真空干燥失重法三种方法,考察各实施例和对比例的水分结果,具体如下:1、热重分析法热重分析是样品在高温状态下分解前的失重,是验证结晶水或吸附水的重要方法,本发明人分别对各实施例和对比例制备的马来酸氯苯那敏化合物进行了热重分析,结果汇总如下:实施例热重法失重(%)实施例10.46实施例20.47实施例30.46对比例10.34结果,实施例1~3制备的1/10水马来酸氯苯那敏化合物失重与含有的1/10个水的结果(理论值0.45%)基本一致;对比例1制备的马来酸氯苯那敏化合物失重与无水物理论值差别不大。推断本发明实施例1~3制备的马来酸氯苯那敏化合物所含水为结晶水,对比例1制备的马来酸氯苯那敏化合物所含水为吸附水。2、60℃热稳定性10天将本发明实施例制备的1/10水马来酸氯苯那敏化合物和对比例1制备的马来酸氯苯那敏化合物分别置于60℃烘箱中10天,分别于0、10天用卡尔费休法检测水分,结果如下:实施例0天(%)10天(%)实施例10.470.46实施例20.470.46实施例30.480.46对比例10.360.08结果,高温60℃放置10天,实施例1~3制备的1/10水马来酸氯苯那敏化合物水分基本没有明显变化,对比例1制备的马来酸氯苯那敏化合物水分明显降低,推断本发明实施例1~3制备的马来酸氯苯那敏化合物所含水为结晶水,对比例1制备的马来酸氯苯那敏化合物所含水为吸附水。3、冷冻真空干燥10小时将本发明实施例1~3制备的1/10水马来酸氯苯那敏化合物和对比例1制备的马来酸氯苯那敏化合物分别置于-35℃冷冻干燥机中抽真空16小时,分别于0、16小时用卡尔费休法检测水分,结果如下:实施例0小时(%)16小时(%)实施例10.470.46实施例20.470.47实施例30.480.47对比例10.360.09结果,低温-35℃冷冻真空干燥16小时,实施例1~3制备的1/10水马来酸氯苯那敏化合物水分基本没有明显变化,对比例1制备的马来酸氯苯那敏化合物水分明显降低,推断本发明实施例1~3制备的马来酸氯苯那敏化合物所含水为结晶水,对比例1制备的马来酸氯苯那敏化合物所含水为吸附水。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1