一种高纯度直链全氟辛酸(L-PFOA)的制备方法与流程
一种高纯度直链全氟辛酸(l-pfoa)的制备方法
技术领域
1.本发明涉及化工产品技术领域,尤其是涉及一种高纯度l-pfoa的制备方法。
背景技术:
2.氟化有机物在工业生产和生活消费领域有着广泛的应用。全氟辛酸(pentadecafluorooctanoic acid,pfoa)及其盐类就是其中一类重要的氟化有机物。它性质稳定、疏油、疏水性,被广泛用于纺织品、皮革品表面防污处理剂;用于生产涂料、泡沫灭火剂和农药等;用于粘合剂、医药产品、阻燃剂、石油及矿业产品,甚至用于纸制食品包装材料和不粘锅。但同时,pfoa作为目前世界上发现的最难降解的有机污染物之一,具有半挥发性,可以长距离转运,长时间在环境中存留,并通过食物链逐级放大,对健康和生态产生了严重影响。
3.目前,有两个问题:
4.第一,通过电化学法生产的pfoa产品直接用于工业和生活产品的生产。在生产、运输和使用前后就会向环境中排放pfoa或相关前体,这就导致土壤、水体、生物等样品中存在多种同分异构体混合物形式的pfoa;且不同污染源、不同区域样品中的pfoa种类和含量都不可控制,差异性非常大。因此,以pfoa为研究对象做毒理学等研究,是从实际出发的最好方案。以pfoa同分异构体单体为研究对象时,结果表明:pfoa同分异构体结构不同,性质也不同,导致在污染物中存在分布差异。l-pfoa(linear-pfoa)为直链全氟辛酸,b-pfoa(branch-pfoa)为支链全氟辛酸,例如:l-pfoa分子结构对称性优于b-pfoa,疏水性较强,更容易与沉积物结合;b-pfoa则更容易在水体中富集。随着时间的推移,这种分布差别也逐渐变大。由上可知,由于结构和性质的差异,分别以pfoa同分异构体单体纯品为研究对象,进行毒理学等评价更具有科学性。
5.第二,pfoa同分异构体分离和检测困难。通过电化学法生产的pfoa,其同分异构体数量多,又有疏水、疏油的特性,在紫外、二极管阵列等检测器上均无响应,造成了pfoa同分异构体分离和检测的困难。光谱法可以实现pfoa总量检测,但是不能区分同分异构体。色谱技术与多种检测器联用以及新型分离材料的迅猛的发展,使得pfoa同分异构体的分离和检测取得了重大进展,但是能够检测到的pfoa同分异构体数量和理论值相比还有很大差距,同时,稍许的差异就可能造成结果不准确,导致错误判定。因此,为了确保方法的稳定性、准确性和数据的溯源性,更需用pfoa同分异构体单体对照品和标准物质作对照和质控。l-pfoa作为含量最高pfoa同分异构体,其高纯度对照品和标准物质的研制对pfoa的准确检测具有十分重要的意义。
6.与检测不同,制备高纯度的pfoa同分异构体单体要克服分离条件苛刻,检测器可选性少的困难;同时,还要在浓缩、干燥等各个环节确保pfoa的稳定性,从而达到对照品和标准物质研制的要求。因此,高纯度pfoa同分异构体单体的制备技术难度远高于其他物质。
7.目前,我国关于l-pfoa和b-pfoa研究的报道较少,pfoa同分异构体单体制备方法尚未见报道,相关标准物质也没有上市。涉及到pfoa的研究和应用都必须从外国购置高纯
度对照品或标准物质,这就受到国际关系、价格波动、时间周期等多种因素的影响。因此,急需自主研发高纯度l-pfoa的制备方法,以满足pfoa毒性研究和相关检测的需求。
8.应该注意,上面对技术背景的介绍只是为了方便对本发明的技术方案进行清楚、完整的说明,并方便本领域技术人员的理解而阐述的。不能仅仅因为这些技术方案在本发明的技术背景部分进行了阐述而认为上述技术方案为本领域技术人员所公知。
技术实现要素:
9.本发明的目的在于提供一种快速、高效的高纯度l-pfoa的制备方法,能够提升制备过程的准确度,确保实验成功;缩短实验周期,提高实验效率,满足准确检测的需求,具有重要的研究意义和使用价值。
10.为实现上述目的,本发明提供了以下技术方案:
11.本发明提供的高纯度l-pfoa的制备方法,包括:
12.分离阶段和浓缩干燥阶段;
13.所述分离阶段包括:
14.步骤一,选取固体pfoa配制成pfoa甲醇溶液;
15.步骤二,利用配备蒸发光散射检测器的制备型高效液相色谱仪对所述pfoa甲醇溶液进行分离,并收集l-pfoa。
16.所述浓缩干燥阶段包括:
17.步骤三,对经过所述步骤二收集到的l-pfoa收集液进行浓缩干燥处理,得到高纯度l-pfoa。
18.可选地,所述固体pfoa中l-pfoa的质量百分含量为90~98%;可选地,所述pfoa甲醇溶液浓度为5~20mg/ml。
19.可选地,所述浓缩干燥处理为:
20.对经过所述步骤二收集到的l-pfoa收集液进行减压蒸馏,将减压蒸馏后的浓缩液经过冷冻干燥处理,得到高纯度的l-pfoa固体。
21.可选地,利用旋转蒸发仪对经过所述步骤二收集到的l-pfoa收集液进行减压蒸馏至剩余液体不超过2ml时停止,将减压蒸馏后的浓缩液转移至-20℃环境冷冻成冰后,通过冷冻干燥仪干燥至全部变成白色粉末,得到的为l-pfoa冻干粉固体。
22.可选地,还包括鉴定阶段;
23.所述鉴定阶段包括:
24.步骤四,利用高效液相色谱-质谱法对所述高纯度l-pfoa进行定量分析;包括:
25.根据质谱峰面积归一化法分析数据,计算得到所述高纯度l-pfoa的质量百分含量w
i
%;
26.对w
i
%的值进行判定;
27.如果w
i
%≥98%,则所述l-pfoa为高纯度l-pfoa;否则,将所述l-pfoa重新进入分离阶段和浓缩干燥阶段,重复操作,直至满足w
i
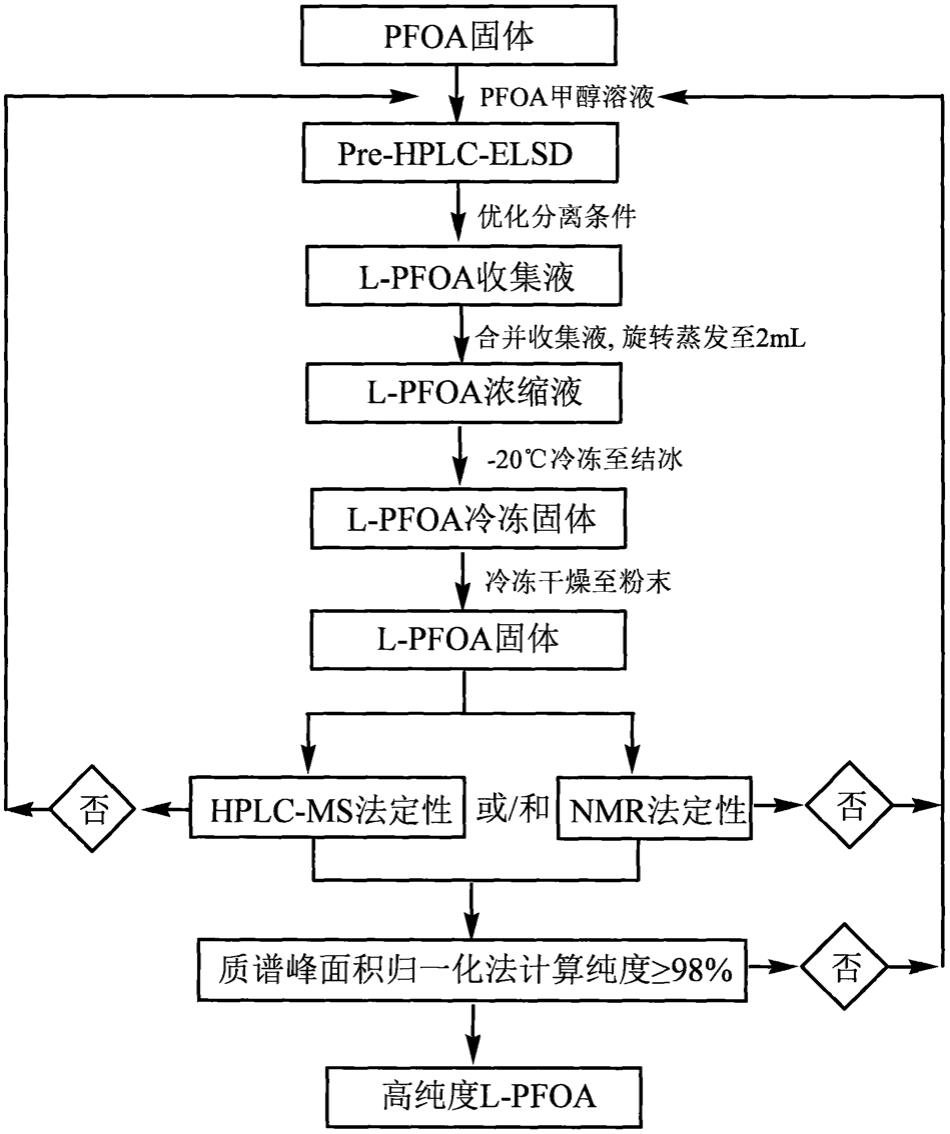
%≥98%。
28.可选地,所述鉴定阶段还包括:
29.在所述步骤四之前,利用高效液相色谱-质谱法对所述高纯度l-pfoa进行定性分析;如果定性分析的结果为否定,则重复分离阶段和浓缩干燥阶段的操作,直到定性分析的
结果为肯定。
30.可选地,所述鉴定阶段还包括:
31.在所述步骤四之前,利用核磁共振波谱的
19
f谱对所述高纯度l-pfoa进行定性分析;如果定性分析的结果为否定,则重复分离阶段和浓缩干燥阶段的操作,直到定性分析的结果为肯定。
32.可选地,步骤二中l-pfoa收集的收集条件为:所述制备型高效液相色谱仪采用氟代辛基键合而成的反相液相色谱柱,色谱柱填料的粒径为5μm,填料的孔径为利用保留时间定性l-pfoa的色谱峰,在检测器出现目标物峰信号时开始进行收集,目标物峰无信号后停止收集。
33.可选地,步骤二中的l-pfoa收集为等度淋洗,对l-pfoa进行反复收集,合并收集液进行所述步骤三。
34.可选地,所述高效液相色谱-质谱法的具体条件为:采用氟代辛基键合而成的反相液相色谱柱,色谱柱填料的粒径为3μm,色谱柱的长度为150mm,色谱柱的内径为2.1mm,填料的孔径为流动相由a和b两相组成,其中,所述a相为甲醇,所述b相为60mmol/l氨水和20mmol/l甲酸水溶液的混合液;采用梯度淋洗。
35.本发明提供的技术方案中,利用蒸发光散射检测器与制备型高效液相色谱仪联用(pre-hplc-elsd)的方式分离、收集目标物l-pfoa;将高效液相色谱分离能力强,制备量大,流动相组成灵活多样,蒸发光散射检测器对l-pfoa有响应的优势结合,通过优化实验条件,解决pfoa同分异构体分离、检测和制备的难题。
36.另外,通过在制备型高效液相色谱系统中配置分流装置,合理控制分流比,实现在线收集过程的可视化,实时监测分离情况,精准控制pfoa收集的起始点,保证收集方法的稳定性。
37.同时,采用高效液相色谱-质谱法(hplc-ms)和核磁共振波谱法(nmr)表征各步实验效果,显著提升制备过程的准确度和成功率,缩短实验周期,提高实验效率,保证了纯化产品的质量。
38.本发明提供的是一种快速、高效的高纯度l-pfoa的制备方法,能够高效的制备出毫克级纯度满足需要的l-pfoa产品(纯度≥98%),获得的高纯度l-pfoa将促进毒理学研究和标准物质研制;有助于填补此项标准物质的空缺,满足准确检测的需求;为其他全氟化合物高纯度单体的研制提供思路。因此,具有重要的研究意义和实用价值。
附图说明
39.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
40.图1是高纯度l-pfoa的制备流程图;
41.图2是pre-hplc-elsd连续制备l-pfoa色谱图;
42.图3是制备的高纯度l-pfoa
19
f核磁共振波谱图;
43.图4是hplc-ms表征pfoa粗品的总离子流图;
44.图5是hplc-ms表征制备的高纯度l-pfoa总离子流图。
具体实施方式
45.为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的技术方案进行详细的描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本发明所保护的范围。
46.高纯度l-pfoa的制备流程图如图1所示,具体的制备方法如下所述,包括:
47.分离阶段和浓缩干燥阶段;
48.分离阶段包括:
49.步骤一,选取固体pfoa配制成pfoa甲醇溶液;
50.步骤二,对pfoa甲醇溶液进行制备型高效液相色谱-蒸发光散射法分离,并收集l-pfoa;
51.浓缩干燥阶段包括:
52.步骤三:经过步骤二收集到的l-pfoa收集液进行浓缩干燥处理,得到l-pfoa。
53.步骤一中,pfoa甲醇溶液浓度为5~20mg/ml之间,能够保证样品溶液浓度合理且不会有沉淀现象发生,本实施例中采用的固体pfoa为市售pfoa对照品(质量百分含量95%),配制成15mg/ml甲醇溶液。
54.步骤二中,对pfoa甲醇溶液进行高效液相色谱-蒸发光散射法分离,并收集l-pfoa,分离效果如图2所示;具体实验条件如下:
55.色谱条件:waters 1525高效液相色谱泵,waters 2707自动样器,分流器(1∶15)(美国waters公司);氟代辛基键合而成的反相液相色谱柱fluorosep-rp octyl(5μm,250mm
×
10mm,es industries),72%a(a相为甲醇,b相为0.01%乙酸的水溶液),等度淋洗,流速5ml/min;进样量150μl。elsd-lti ii型蒸发光检测器(日本shimadzu公司):漂移管温度40℃,压力385kpa,信号放大倍数6,载气为高纯氮气。利用保留时间定性l-pfoa的色谱峰,在监测器出现峰信号时开始对目标物进行收集,目标物峰无信号后停止收集。按照上述步骤反复收集,将收集到的l-pfoa溶液合并。
56.于本发明的具体实施例中,分流比为15∶1。具体是指:流动相从色谱柱中出来之后,利用分流器(类似一个三通装置)将其按照体积比15∶1的比例进行分配。其中,1份进入检测器进行检测,用于确定收集窗口的时间;另外15份进行收集,作为下一步实验的材料。于本发明的具体实施例中,相对于传统的收集方法,增设有工作站,工作站的设置能够实时监测分离过程,通过合理控制分流比,实现在线收集过程的可视化,通过工作站实时监测分离情况,筛选得到收集的起始点,做到准确收集,保证了收集的稳定性。
57.于本发明的具体实施例中,浓缩干燥处理为:
58.利用旋转蒸发仪对经过步骤二收集到的l-pfoa收集液进行减压蒸馏至剩余液体不超过2ml时停止,将减压蒸馏后的浓缩液转移至样品瓶和-20℃环境冷冻成冰后,经过冷冻干燥处理得到纯化后的l-pfoa白色粉末状固体。
59.利用旋转蒸发仪对经过步骤二收集到的l-pfoa收集液进行减压蒸馏至剩余液体不超过2ml时停止,但是这只是本发明的一种具体实施例,并不局限于2ml,还可以为其他能
够满足实验需求的体积,均是可以根据实际操作需求来进行实时调整的。
60.为了判断制备得到的产品是否为l-pfoa及其纯度是否满足要求,于本发明的具体实施例中,还包括鉴定阶段,具体分为定性和定量分析阶段;
61.为了确保定性分析的可靠性,在定性分析阶段分别采用高效液相色谱-质谱法和核磁共振波谱法两种手段相互佐证,具体包括:
62.利用高效液相色谱-质谱法对所述高纯度l-pfoa进行定性分析;如果定性分析的结果表明收集的组分不是l-pfoa,则重复分离阶段和浓缩干燥阶段的操作,直到定性分析的结果为肯定。
63.具体的实验条件如下:
64.色谱条件:acquity超高效液相色谱仪,xevo tq串联质谱仪,masslynx 4.1工作站(美国waters公司);氟代辛基键合而成的反相液相色谱柱fluorosep-rp octyl(3μm,150mm
×
2.1mm,es industries),流动相由a和b两相组成(a相为甲醇,b相为60mmol/l氨水+20mmol/l甲酸水溶液),流速0.2ml/min,梯度淋洗:a:0-3min,50%;3-10min,50%-64%;10-20min,64%-66%;20-30min,66%-70%;30-100min,70%-78%。质谱条件:电喷雾负离子模式;毛细管电压:3.0kv;萃取电压5.0v;离子源温度:150℃;脱溶剂气温度350℃;脱溶剂气体流速650l/h;锥孔气流速20l/h;母离子:412.09;锥孔电压40v。于本发明的实施例中,具体指:质谱检测l-pfoa时候,l-pfoa的质荷比为412.09,如果能够检测到这个质荷比,就认为是l-pfoa。
65.另一种定性方法,则是利用核磁共振波谱的
19
f谱对所述高纯度l-pfoa进行定性分析;如果定性分析的结果为否定,则重复分离阶段和浓缩干燥阶段的操作,直到定性分析的结果为肯定。
66.本实施例中,纯化后l-pfoa的核磁共振
19
f谱如图3所示,可以看到纯化产物正是期望的l-pfoa,且杂质峰非常少。
67.具体的实验条件如下:
68.仪器型号:agilent dd2 600m nmr;采用重水为氘代溶剂;观测频率564mhz;观测核为
19
f;脉冲延迟时间1s;采样次数8次。
69.定量分析阶段具体包括:
70.利用高效液相色谱-质谱法对所述高纯度l-pfoa进行定量分析;
71.具体的实验条件如下:
72.色谱条件:acquity超高效液相色谱仪,xevo tq串联质谱仪,masslynx 4.1工作站(美国waters公司);氟代辛基键合而成的反相液相色谱柱fluorosep-rp octyl(3μm,150mm
×
2.1mm,es industries),流动相由a和b两相组成(a相为甲醇,b相为60mmol/l氨水+20mmol/l甲酸水溶液),流速0.2ml/min,梯度淋洗:a:0-3min,50%;3-10min,50%-64%;10-20min,64%-66%;20-30min,66%-70%;30-100min,70%-78%。质谱条件:电喷雾负离子模式,sir模式;毛细管电压:3.0kv;萃取电压5.0v;离子源温度:150℃;脱溶剂气温度350℃;脱溶剂气体流速650l/h;锥孔气流速20l/h;母离子:412.09;锥孔电压40v。
73.采用质谱峰面积归一化法分析数据,计算得到l-pfoa纯度(质量百分含量),表示如下:
74.l-pfoa的纯度为w
i
%;
[0075][0076]
m
i
、a
i
、w
i
%分别为组分l-pfoa的质量、峰面积、质量分数,为组分l-pfoa的定量校正因子(也可叫做相对峰值面积百分数),根据多次实验结果,发现单一峰值面积较大,其他成分峰值可以忽略不计,因此,于本发明的具体实施例中,定量校正因子取值为1;
[0077]
比较w
i
%与98%的大小;
[0078]
如果w
i
%≥98%,则l-pfoa为高纯度l-pfoa;否则,将l-pfoa重新进入分离阶段和浓缩干燥阶段,重复操作,直至满足w
i
%≥98%。
[0079]
本实施例中,图4和图5分别是hplc-ms表征的纯化前l-pfoa粗品和纯化后l-pfoa的总离子流图,由图可知,纯化后的l-pfoa杂质峰明显减少。实验得到的测试结果为:纯化后l-pfoa的纯度为w
i
%=98.3%,满足w
i
%≥98%的要求。
[0080]
质谱峰面积归一化法是同内标法、外标法并列的一种化学计算方法。当没有标准物质或者对照品的时候,可以采用这种方法计算纯度。具体如下:面积归一法适用于复杂样品组分的含量测定,前提是样品中所有组分全部流出色谱柱并在色谱图上都出现色谱峰。因此,进样量的准确性和操作条件的变动对测定结果影响不大。
[0081]
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1