一种含有吡啶并三氮唑及衍生物受体结构单元的可见光延迟荧光材料及应用的制作方法
本发明涉及可见光延迟荧光材料领域,尤其涉及一种含有吡啶并三氮唑及衍生物受体结构单元的可见光延迟荧光材料及应用。
背景技术:
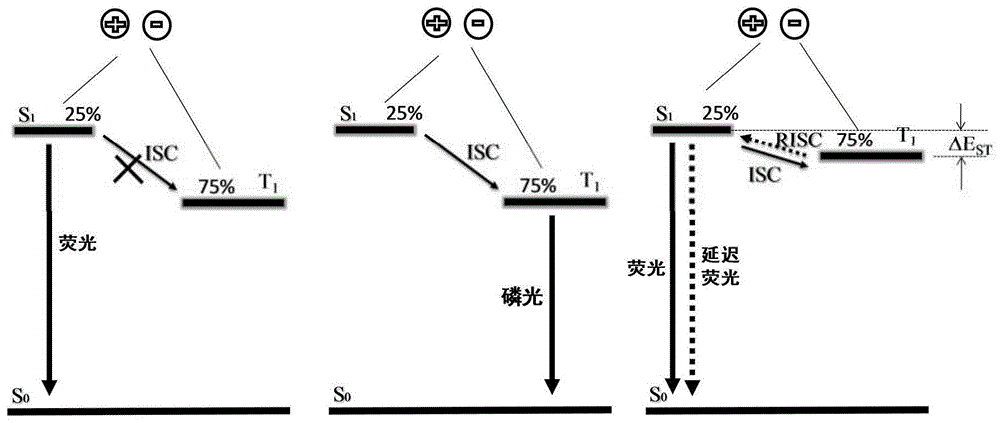
:oled即有机发光二极管(organiclight-emittingdiode)或有机发光器件(organiclight-emittingdevice),是一种无需背光源,自主发光材料;具有全固态、宽视角、耐低温、色彩逼真、驱动电压低、响应速度快、对比度及清晰度高、超薄、易于柔性显示等优点,亦能以成本低廉的玻璃、柔性金属和塑料为基板;此外,还具有高能效、低能耗、材料来源广泛、生产工艺简单、平面发光、可大面积生产等诸多优势。oled作为新一代的照明与显示技术,已应用于手机、平板、相机、电视、电脑、检测仪器等产品,且在航空航天、平面固态照明等领域有着潜在的应用前景。发光材料是oled器件的核心部分,有机发光材料按发光原理大致可以分为荧光材料、磷光材料和热活化延迟荧光(tadf)材料。如附图1所示,荧光材料为最早应用的第一代oled材料,受限于电子自旋统计只能利用单线态激子发光,电致发光器件的内量子效率最高达25%。1988年,美国普林斯顿大学的forrest教授报道了室温下金属有机铂配合物的磷光电致发光现象,器件内量子效率能达到100%。虽然截至目前金属有机磷光材料已取得了长足的发展,红光和绿光金属铱配合物磷光材料已实现在商业化电子产品中的应用,但是磷光材料由于要使用稀有且昂贵的贵金属致使其价格十分昂贵,且资源及其有限。热活化延迟荧光(tadf)材料通过三线态激子的反转换实现100%的电致发光效率,其发光效率可以和磷光材料的相媲美,且可避免贵金属的使用。因此,开发新型、且可用于oled器件的热活化延迟荧光材料对我国oled产业的发展具有十分重要的意义。技术实现要素:为了开发更多种类、更高性能的热活化延迟荧光材料,本发明提供了一种含有吡啶并三氮唑及衍生物受体结构单元的可见光延迟荧光材料及应用,本发明提出的热活化延迟荧光材料通过精巧的分子设计,采用给体-受体分子体系,并且严格调控给体和受体的分子结构,使分子的最高占有轨道(homo)和最低非占有轨道(lumo)能够实现空间上的分离。而空间上分离的homo和lumo可促进激发态分子由lumo到homo之间的电荷转移(分子内电荷转移),进而辐射发光;同时亦可使低激发单线态(s1)和最低激发三线态(t1)之间的能级差(δest)变小(≤0.2ev),发生快速的反系间窜越(risc),进而实现延迟荧光发光。为了实现上述目的,本发明采用如下技术方案:一种含有吡啶并三氮唑及衍生物受体结构单元的可见光延迟荧光材料,包含下述通式(1)所述的化合物:式(1)中,x为氧原子、硫原子或-c(rxry)-,所述的rx、ry、ra1、rb1、rc1和rd1各自独立地为氢原子或者取代基,m1、n1、o1和p1代表氢原子或者取代基的个数;相邻的取代基可以相连形成稠环,所述稠环还可以与其他环稠合。作为本发明的优选实施方式,所述的取代基选自c1-c24的烷基、c1-c24的烷氧基、c3-c24的环烷基、c1-c24的醚、c1-c24的杂环基、c1-c24的芳基、c4-c24的芳氧基、卤素、单或二烷基氨基、单或二芳基氨基、氰基或其组合。进一步优选为c1-c6的烷基、c1-c6的烷氧基、c3-c6的环烷基、c1-c6的杂环基、苯基、苯氧基。作为本发明的优选实施方式,所述的m1、n1、o1和p1为0-4的整数,进一步优选为0、1或2。作为本发明的优选实施方式,所述的rc1和rd1为氢原子。作为本发明的优选实施方式,所述的ra1为氢原子。作为本发明的优选实施方式,所述的通式(1)具体为下述通式(ⅰ)至(ⅳ)中的任意一种,其中,x为氧原子、硫原子或-c(rxry)-,所述的rx、ry各自独立地为c1-c6的烷基、c1-c6的烷氧基、c3-c6的环烷基、c1-c6的杂环基、苯基、苯氧基。作为本发明的优选实施方式,通式(1)所述的化合物为下述结构式中的任意一种,作为本发明的优选实施方式,所述的荧光材料发射延迟荧光。本发明的另一目的在于提供一种上述的含有吡啶并三氮唑及衍生物受体结构单元的可见光延迟荧光材料在有机电致发光器件中的应用,所述可见光延迟荧光材料作为有机电致发光器件中的发光层材料。与现有技术相比,本发明的有益效果在于:(1)本发明采用吡啶并三氮唑及其衍生物作为受体,苯噁嗪、苯噻嗪或哑啶及其衍生物作为给体,二者之间通过苯环或取代的苯环链接,通过受体及给体结构调控,从而调节给体-苯环-受体之间的二面角。在受体结构一样的情况下,调控给体的结构对受体/苯环之间的二面角影响不大,但是苯环/给体之间的二面角则受给体结构影响巨大,本发明通过将苯噁嗪、苯噻嗪或哑啶及其衍生物作为给体,由于其和苯环之间的位阻大,使苯环/给体之间的二面角高达80°以上,苯环/给体之间几乎处于垂直的空间结构,有效减少了苯环和给体之间的共轭,实现了分子homo和lumo空间上的有效分离,而空间上分离的homo和lumo可使s1和t1之间的能级差(δest)变小,当δest≤0.2ev,可发生快速的反系间窜越(risc),进而实现延迟荧光发光,本发明中的化合物δest均在0.1ev左右,因此基于本发明的延迟荧光材料具有较高的发光效率。(2)本发明通过调控给体的结构,进而调节其电性和给电子能力,可对荧光材料的最高占有分子轨道(homo)和最低未占有分子轨道(lumo)的轨道能级进行有效调控,使得可调控材料发光颜色处于400-700nm的可见光区。通过对荧光材料掺杂的dpepo薄膜的瞬态发光光谱的研究,证明本发明提出的荧光材料显示出两个辐射跃迁发光寿命,分别为短寿命的s1→s0跃迁和长寿命的t1→s1跃迁,证明延迟荧光的发生,且光致发光量子效率较高,可强烈发光。附图说明图1为电致荧光磷光和延迟荧光发光的机理图。其中s0为材料分子基态,s1为材料分子最低激发单线态,t1为材料分子最低激发三线态,isc为系间窜越,risc为反系间窜越,δest为s1和t1之间的能级差。图2为本发明的化合物通式结构。图3为通过密度泛函理论(dft)计算得到的1-o、1-s和1-c的homo及lumo轨道分布。图4为通过密度泛函理论(dft)计算得到的3-o、3-s和3-c的homo及lumo轨道分布。图5为通过密度泛函理论(dft)计算得到的1-cz、1-nph2和1-czph的homo及lumo轨道分布。图6为发光材料1-o室温下在不同溶液中的发射光谱,其中toluene为甲苯,ea为乙酸乙酯,thf为四氢呋喃,dcm为二氯甲烷,ch3cn为乙腈;各溶剂后括号内的数字为其介电常数。图7为发光材料1-s室温下在不同溶液中的发射光谱,其中toluene为甲苯,ea为乙酸乙酯,thf为四氢呋喃,dcm为二氯甲烷,ch3cn为乙腈;各溶剂后括号内的数字为其介电常数。图8为发光材料1-c室温下在不同溶液中的发射光谱,其中toluene为甲苯,ea为乙酸乙酯,thf为四氢呋喃,dcm为二氯甲烷,ch3cn为乙腈;各溶剂后括号内的数字为其介电常数。图9为发光材料2-o室温下在不同溶液中的发射光谱,其中toluene为甲苯,ea为乙酸乙酯,thf为四氢呋喃,dcm为二氯甲烷,ch3cn为乙腈;各溶剂后括号内的数字为其介电常数。图10为发光材料1-czph室温下在不同溶液中的发射光谱,其中toluene为甲苯,ea为乙酸乙酯,thf为四氢呋喃,dcm为二氯甲烷,ch3cn为乙腈;各溶剂后括号内的数字为其介电常数。图11为发光材料3-cz室温下在不同溶液中的发射光谱,其中toluene为甲苯,ea为乙酸乙酯,thf为四氢呋喃,dcm为二氯甲烷,ch3cn为乙腈;各溶剂后括号内的数字为其介电常数。图12为发光材料3-cztbu室温下在不同溶液中的发射光谱,其中toluene为甲苯,ea为乙酸乙酯,thf为四氢呋喃,dcm为二氯甲烷,ch3cn为乙腈;各溶剂后括号内的数字为其介电常数。图13为发光材料3-czph室温下在不同溶液中的发射光谱,其中toluene为甲苯,ea为乙酸乙酯,thf为四氢呋喃,dcm为二氯甲烷,ch3cn为乙腈;各溶剂后括号内的数字为其介电常数。图14为发光材料1-o、1-s、2-o、2-cztbu、2-cztph和2-czt室温下在其掺杂的二[2-((氧代)二苯基膦基)苯基]醚(dpepo)薄膜的瞬态发光光谱。图15为有机发光元件的结构示意图。具体实施方式下面通过具体实施例对本发明作进一步说明,但本发明的保护范围并不仅限于此。除非另有说明,以下试验中所涉及到的所有商业试剂购买后直接使用,没有进一步纯化。核磁共振氢谱和碳谱均在氘代氯仿(cdcl3)溶液中测得,氢谱使用400或500兆赫兹的核磁共振谱仪,碳谱使用100或126兆赫兹的核磁共振谱仪,化学位移以四甲基硅烷(tms)或残留溶剂为基准。用氘代氯仿(cdcl3)作溶剂,氢谱和碳谱分别以tms(δ=0.00ppm)和cdcl3(δ=77.00ppm)作为内标。以下缩写(或组合)用于解释氢谱峰:s=单峰,d=双重峰,t=三重峰,q=四重峰,p=五重峰,m=多重峰,br=宽峰。高分辨质谱在赛默飞世尔科技有限公司的ltqftultra质谱仪上测得,样品电离模式为电喷雾电离。如图2,本发明提供了一种含有吡啶并三氮唑及衍生物受体结构单元的可见光延迟荧光材料,包含下述通式(1)所述的化合物:式(1)中,ra1、rb1、rc1和rd1各自独立地为氢原子或者c1-c24的烷基、c1-c24的烷氧基、c3-c24的环烷基、c1-c24的醚、c1-c24的杂环基、c1-c24的芳基、c4-c24的芳氧基、卤素、单或二烷基氨基、单或二芳基氨基、氰基或其组合,其中相邻的两个取代基可以稠合成环。m1、n1、o1和p1代表氢原子或者取代基的个数,优选为0-4的整数,进一步优选为0、1或2。x为o、s或-c(rxry)-,其中所述的rx和ry选自c1-c24的烷基、c1-c24的烷氧基、c3-c24的环烷基、c1-c24的醚、c1-c24的杂环基、c1-c24的芳基、c4-c24的芳氧基或其组合,优选为c1-c6的烷基、c1-c6的烷氧基、c3-c6的环烷基、c1-c6的杂环基、苯基、苯氧基。在本发明的一个具体实施中,通式(1)所述的化合物为下述结构式中的任意一种,根据本发明的一个实施方式的化合物可以利用下述制备方法来制备。下述实施例中,虽然记载了代表性的例子,然而,不解释为限制本发明,可以更具需要追加或删除取代基,也可以改变取代基的位置。实施例1:化合物1-o的合成路线如下:1a的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入对溴苯甲醛(183mg,1.00mmol,1.0当量),吩噁嗪(220mg,1.2mmol,1.2当量),醋酸钯(7mg,0.03mmol,3mol%),碳酸钾(415mg,3.00mmol,3当量),然后抽换氮气三次,在氮气保护下加入三叔丁基膦(6.2mg,0.08mmol,8.0mol%)和甲苯(4ml)。该混合物在120℃油浴中搅拌反应12小时,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的水,并用乙酸乙酯萃取。水层用乙酸乙酯萃取两次。合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=20:1,得到中间体1a,淡黄色固体273mg,收率95%。1hnmr(500mhz,cdcl3):δ5.96(dd,j=8.0,1.5hz,2h),6.62(td,j=8.5,2.0hz,2h),6.68-6.74(m,4h),7.54-7.57(m,2h),8.10-8.13(m,2h),10.11(s,1h)。1-o的合成:在带有磁力搅拌子的反应瓶中加入中间体1a(287mg,1.00mmol,1.0当量),2-肼基吡啶(109mg,1.00mmol,1.0当量),然后抽换氮气三次,在氮气的保护下加入乙醇(8ml),在80℃的油浴锅中搅拌反应2小时。薄层色谱监测至原料反应完毕,冷却至室温。反应液移至单口瓶,真空减压蒸馏除去乙醇溶液,得到中间体1b直接进行下一步。在单口瓶中加入碘单质(305mg,1.20mmol,1.2当量),碳酸钾(415mg,3.00mmol,3.0当量)及二氯甲烷溶液(10ml),在室温下搅拌反应15小时,反应产生大量的黑色固体。反应液用硫代硫酸钠溶液洗涤,并且水相用二氯甲烷溶液萃取两遍,合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=5:1-1:1,得到1-o,淡黄色固体118mg,两步的收率53%。1hnmr(600mhz,cdcl3):δ6.02(d,j=7.8hz,2h),6.37(t,j=7.8hz,2h),6.69(t,j=7.2hz,2h),6.73(d,j=7.8hz,2h),7.00(t,j=7.2hz,1h),7.38-7.41(m,1h),7.61(d,j=7.8hz,2h),7.93(d,j=9.0hz,1h),8.10(d,j=8.4hz,2h),8.41(d,j=6.6hz,1h)。13cnmr(600mhz,cdcl3):δ113.25,114.63,115.64,116.96,121.74,122.53,123.27,126.76,127.36,30.78,132.00,133.85,140.94,143.92,145.90,150.65。c24h16n4nao的计算值399.1216,实测值399.1211。实施例2:化合物1-s的合成路线如下:2a的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入对溴苯甲醛(925mg,5.00mmol,1.0当量),吩噻嗪(996mg,5.00mmol,1.2当量),醋酸钯(34mg,0.15mmol,3mol%),碳酸钾(2.07g,15.00mmol,3.0当量),然后抽换氮气三次,在氮气保护下加入三叔丁基膦(81mg,0.4mmol,8.0mol%)和甲苯/二氧六环(3ml/3m)。该混合物在110℃油浴中搅拌反应15小时,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的水,并用乙酸乙酯萃取。水层用乙酸乙酯萃取两次。合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=20:1-101,得到中间体2a,浅棕固体1.36g,收率90%。1hnmr(600mhz,cdcl3):δ7.15-7.19(m,4h),δ7.26-7.29(m,4h),7.42(d,j=7.8hz,2h),7.75(d,j=8.4hz,2h),9.85(s,1h)。1-s的合成:在带有磁力搅拌子的反应瓶中加入中间体2a(607mg,1.00mmol,1.0当量),2-肼基吡啶(218mg,2.00mmol,1.0当量),然后抽换氮气三次,在氮气的保护下加入乙醇(10ml),在80℃的油浴锅中搅拌反应54.5小时。薄层色谱监测至原料反应完毕,冷却至室温。反应液移至单口瓶,真空减压蒸馏除去乙醇溶液,得到中间体2b直接进行下一步。在单口瓶中加入碘单质(609mg,2.40mmol,1.2当量),碳酸钾(829mg,6.00mmol,3.0当量)及二氯甲烷溶液(10ml),在室温下搅拌反应11小时,反应产生大量的黑色固体。反应液用硫代硫酸钠溶液洗涤,并且水相用二氯甲烷溶液萃取两遍,合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=5:1-1:1,得到1-s,淡黄色固体569mg,两步的收率73%。1hnmr(500mhz,cdcl3):δ6.63(dd,j=8.0,1.5hz,2h),6.91(td,j=7.0,1.5hz,1h),6.96(td,j=7.5,1.5hz,2h),7.03(td,j=7.5,1.5hz,2h),7.18(dd,j=8.0,1.5hz,2h),7.32(ddd,j=9.5,6.5,1.0hz,1h),7.50-7.53(m,2h),7.86(d,j=9.0hz,1h),7.96-7.99(m,2h),8.35(d,j=7.0hz,1h)。13cnmr(500mhz,cdcl3):δ114.37,116.80,119.22,122.58,123.73,124.23,124.44,126.98,127.16,127.28,127.40,130.07,143.08,144.12,146.13,150.52。c24h16n4nas的计算值415.0988,实测值415.0982。实施例3:化合物1-c的合成路线如下:3a的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入对溴苯甲醛(925mg,5.00mmol,1.0当量),9,9-二甲基哑啶(1.26g,5.00mmol,1.2当量),醋酸钯(34mg,0.15mmol,3mol%),碳酸钾(2.07g,15.00mmol,3当量),然后抽换氮气三次,在氮气保护下加入三叔丁基膦(81mg,0.4mmol,8.0mol%)和甲苯(15ml)。该混合物在110℃油浴中搅拌反应10小时,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的水,并用乙酸乙酯萃取。水层用乙酸乙酯萃取两次。合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=20:1-5:1,得到中间体3a,浅棕固体1.56g,收率99%。1hnmr(600mhz,cdcl3)δ6.80(s,6h),6.33(d,j=8.4hz,2h),6.96-7.01(m,4h),7.48(dd,j=7.2,1.2hz,2h),7.54(d,j=7.8hz,2h),8.12(d,j=7.8hz,2h),10.12(s,1h)。1-c的合成:在带有磁力搅拌子的反应瓶中加入中间体3a(313mg,1.00mmol,1.0当量),2-肼基吡啶(109mg,1.00mmol,1.0当量)然后抽换氮气三次,在氮气的保护下加入乙醇(6ml),在80℃的油浴锅中搅拌反应5.5小时。薄层色谱监测至原料反应完毕,冷却至室温。反应液移至单口瓶,真空减压蒸馏除去乙醇溶液,得到中间体3b直接进行下一步。在单口瓶中加入碘单质(305mg,1.20mmol,1.2当量),碳酸钾(415mg,3.00mmol,3.0当量)及二氯甲烷溶液(10ml),在室温下搅拌反应4.5小时,反应产生大量的黑色固体。反应液用硫代硫酸钠溶液洗涤,并且水相用二氯甲烷溶液萃取两遍,合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=1:1,得到1-c,淡黄色固体193mg,两步的收率76%。1hnmr(500mhz,cdcl3):δ1.72(s,6h),6.35(dd,j=8.5,1.5hz,2h),6.95-6.98(m,3h),6.99-7.03(m,2h),7.35(ddd,j=9.0,6.5,1.0hz,1h),7.49(dd,j=8.0,2.0hz,2h),7.59(d,j=8.5hz,2h),7.90(d,j=9.5hz,1h),8.13(d,j=8.5hz,2h),8.44(dd,j=7.0hz,1h)。13cnmr(600mhz,cdcl3):δ35.98,31.17,114.03,114.56116.96,120.94,122.62,125.33,126.40,126.48,127.30,130.26,130.64,132.44,140.54,143.20,146.10,150.65。c27h22n4na的计算值425.1737,实测值425.1732。实施例4:化合物2-o的合成路线如下:4a的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入4-溴-2,6-二甲基苯甲醛(213mg,1.00mmol,1.0当量),吩噁嗪(401g,1.20mmol,1.2当量),醋酸钯(7mg,0.03mmol,3mol%),碳酸钾(415mg,3.00mmol,3当量),然后抽换氮气三次,在氮气保护下加入三叔丁基膦(16mg,0.08mmol,8.0mol%)和甲苯(6ml)。该混合物在110℃油浴中搅拌反应6小时,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的水,并用乙酸乙酯萃取。水层用乙酸乙酯萃取两次。合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=1:0-20:1,得到中间体4a,金黄色固体272mg,收率86%。1hnmr(500mhz,cdcl3):δ2.66(s,6h),5.98(dd,j=8.0,1.5hz,2h),6.62(td,j=8.0,1.5hz,2h),6.66-6.67(m,4h),7.10(s,2h),10.66(s,1h)。2-o的合成:在带有磁力搅拌子的反应瓶中加入中间体4a(252mg,0.80mmol,1.0当量),2-肼基吡啶(87mg,0.80mmol,1.0当量)然后抽换氮气三次,在氮气的保护下加入乙醇(7ml),在80℃的油浴锅中搅拌反应5.5小时。薄层色谱监测至原料反应完毕,冷却至室温。反应液移至单口瓶,真空减压蒸馏除去乙醇溶液,得到中间体4b直接进行下一步。在单口瓶中加入碘单质(244mg,0.96mmol,1.2当量),碳酸钾(332mg,2.40mmol,3.0当量)及二氯甲烷溶液(8ml),在室温下搅拌反应36小时,反应产生大量的黑色固体。反应液用硫代硫酸钠溶液洗涤,并且水相用二氯甲烷溶液萃取两遍,合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=5:1,得到2-o,淡黄色固体203mg,两步的收率63%。1hnmr(500mhz,cdcl3):δ2.10(s,6h),6.04(d,j=2.0hz,1h),6.05-6.06(m,1h),6.64-6.73(m,6h),6.91(td,j=6.5,0.5hz,1h),7.23(s,2h),7.36(ddd,j=9.5,6.5,1.0hz,1h),7.65(s,1h),7.90(s,1h)。c26h20n4nao的计算值427.1529,实测值427.1521。为了更好地说明问题,本发明也对以下几个对比化合物做了研究。对比实施例5:发光材料1-czph的合成路线如下:3,6-二苯基咔唑的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入3,6-二溴咔唑(9.75g,30.00mmol,1.0当量),苯硼酸(8.78g,72.00mmol,2.4当量),四(三苯基膦)钯(347mg,0.3mmol,10mol%),碳酸钾(20.73g,150.00mmol,3当量),然后抽换氮气三次,在氮气保护下加入甲苯/乙醇/水(30ml/30ml/10ml)。该混合物在80℃油浴中搅拌反应20小时,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的水,并用二氯甲烷两次萃取。合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=20:1-5:1,得到中间体3,6-二苯基咔唑,浅棕固体7.95g,收率83%。5a的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入对溴苯甲醛(555mg,3.00mmol,1.5当量),3,6-二苯基咔唑(639.0g,2mmol,1.0当量),pd2(dba)3(73mg,0.08mmol,4mol%),碳酸钾(829mg,15.00mmol,3当量),然后抽换氮气三次,在氮气保护下加入甲苯(6ml)。该混合物在110℃油浴中搅拌反应20小时,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的水,并用乙酸乙酯萃取,水层用乙酸乙酯萃取两次,合并有机相。无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/二氯甲烷/乙酸乙酯=20:2:1-1:0:0,得到中间体5b,金黄色固体722mg,收率85%。1hnmr(500mhz,cdcl3):δ7.37(t,j=7.5hz,2h),7.50(t,j=7.5hz,4h),7.59(d,j=4.0hz,2h),7.71(dd,j=8.5,2.0hz,2h),7.73-7.75(m,4h),7.86(d,j=8.5hz,2h),8.18(d,j=8.5hz,2h),8.40(d,j=1.5hz,2h),10.14(s,1h)。1-czph的合成:在带有磁力搅拌子的反应瓶中加入中间体5a(424mg,1.00mmol,1.0当量),2-肼基吡啶(109mg,1.00mmol,1.0当量)然后抽换氮气三次,在氮气的保护下加入乙醇(8ml),在80℃的油浴锅中搅拌反应22小时。薄层色谱监测至原料反应完毕,冷却至室温。反应液移至单口瓶,真空减压蒸馏除去乙醇溶液,得到中间体5b直接进行下一步。在单口瓶中加入碘单质(305mg,1.20mmol,1.2当量),碳酸钾(415mg,3.00mmol,3.0当量)及二氯甲烷溶液(10ml),在室温下搅拌反应4.5小时,反应产生大量的黑色固体。反应液用硫代硫酸钠溶液洗涤,并且水相用二氯甲烷溶液萃取两遍,合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=1:1,得到1-czph,淡黄色固体393mg,两步的收率77%。1hnmr(600mhz,cdcl3):δ6.97(t,j=6.6hz,1h),7.35-7.38(m,3h),7.50(t,j=7.2hz,4h),7.58(d,j=8.4hz,2h),7.72(d,j=8.4hz,2h),7.74(d,j=7.8hz,4h),7.80(d,j=8.4hz,2h),7.91(d,j=9.0hz,1h),8.13(d,j=7.8hz,2h),8.42(s,2h),8.44(d,j=7.2hz,1h)。13cnmr(600mhz,cdcl3):δ110.02,114.52,116.88,118.88,122.50,124.30,125.42,125.79,126.68,127.21,127.35,128.77,129.68,134.08,139.32,140.22,141.55,145.95,150.64。c36h24n4na的计算值535.1893,实测值535.1888。对比实施例6:发光材料2-cz的合成路线如下:6a的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入4-溴-2,6-二甲基苯甲醛(526mg,2.00mmol,1.0当量),咔唑(401mg,2.40mmol,1.2当量),醋酸钯(14mg,0.06mmol,3mol%),碳酸钾(829mg,6.00mmol,3当量)然后抽换氮气三次,在氮气保护下加入三叔丁基膦(32mg,0.16mmol,8.0mol%)和甲苯(6ml)。该混合物在110℃油浴中搅拌反应10小时,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的水,并用乙酸乙酯萃取。水层用乙酸乙酯萃取两次。合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=20:1-5:1,得到中间体6a,浅棕色固体578mg,收率99%。1hnmr(500mhz,cdcl3):δ2.73(s,6h),7.25(s,2h),7.31(t,j=7.2hz,2h),7.43(t,j=7.2hz,2h),7.49(d,j=8.4hz,2h),8.13(d,j=7.8hz,2h),10.69(s,1h)。2-cz的合成:在带有磁力搅拌子的反应瓶中加入中间体6a(299mg,1.00mmol,1.0当量),2-肼基吡啶(109mg,1.00mmol,1.0当量),然后抽换氮气三次,在氮气的保护下加入乙醇(6ml),在80℃的油浴锅中搅拌反应6.5小时。薄层色谱监测至原料反应完毕,冷却至室温。反应液移至单口瓶,真空减压蒸馏除去乙醇溶液,得到中间体6b直接进行下一步。在单口瓶中加入碘单(305mg,1.20mmol,1.2当量),碳酸钾(415mg,3.00mmol,3.0当量)及二氯甲烷溶液(10ml),在室温下搅拌反应12小时,反应产生大量的黑色固体。反应液用硫代硫酸钠溶液洗涤,并且水相用二氯甲烷溶液萃取两遍,合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯=1:1,得到2-cz,淡黄色固体350mg,两步的收率90%。1hnmr(500mhz,cdcl3):δ2.16(s,6h),6.91(td,j=6.0,1.0hz,1h),7.32(td,j=8.0,1.0hz,2h),7.36(ddd,j=9.5,6.5,1.5hz,1h),7.47-7.44(m,4h),7.52(d,j=8.5hz,2h),7.72(dt,j=7.0,1.0hz,1h),7.91(dt,j=9.0,1.0hz,1h),8.17(d,j=7.5hz,2h)。13cnmr(600mhz,cdcl3):δ19.83,109.73,114.23,116.69,120.15,120.30,122.19,123.49,124.16,125.97,126.01,127.18,139.73,140.45,141.46,144.86,149.78。c26h21n4的计算值389.1761,实测值389.1775。对比实施例7:发光材料2-cztbu的合成路线如下:7a的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入4-溴-2,6-二甲基苯甲醛(213mg,1.00mmol,1.0当量),3,6-二叔丁基咔唑(401g,1.20mmol,1.2当量),醋酸钯(7mg,0.03mmol,3mol%),碳酸钾(414mg,3.00mmol,3当量)然后抽换氮气三次,在氮气保护下加入三叔丁基膦(16mg,0.08mmol,8.0mol%)和甲苯(6ml)。该混合物在110℃油浴中搅拌反应2天,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的水,并用乙酸乙酯萃取。水层用乙酸乙酯萃取两次。合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚=10:1-5:1,得到中间体7a,浅白色固体663mg,收率81%。1hnmr(500mhz,cdcl3):δ1.47(s,18h),2.72(s,6h),7.35(s,2h),7.44-7.69(m,4h),8.13(d,j=1.5hz,2h),10.67(s,1h)。2-cztbu的合成:在带有磁力搅拌子的反应瓶中加入中间体7a(299mg,1.00mmol,1.0当量),2-肼基吡啶(109mg,1.00mmol,1.0当量)然后抽换氮气三次,在氮气的保护下加入乙醇(6ml),在80℃的油浴锅中搅拌反应6小时。薄层色谱监测至原料反应完毕,冷却至室温。反应液移至单口瓶,真空减压蒸馏除去乙醇溶液,得到中间体7b直接进行下一步。在单口瓶中加入碘单质(305mg,1.20mmol,1.2当量),碳酸钾(415mg,3.00mmol,3.0当量)及二氯甲烷溶液(8ml),在室温下搅拌反应3小时,反应产生大量的黑色固体。反应液用硫代硫酸钠溶液洗涤,并且水相用二氯甲烷溶液萃取两遍,合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/二氯甲烷/乙酸乙酯=1:2:1-1:3:1,得到2-cztbu,淡黄色固体400mg,两步的收率80%。1hnmr(500mhz,cdcl3):δ1.48(s,18h),2.14(s,6h),6.90(td,j=7.0,1.0hz,1h),7.36(ddd,j=9.5,6.5,1.5hz,1h),7.46(d,j=8.5hz,4h),7.51(dd,j=8.5,2.0hz,2h),7.72(td,j=7.0,1.0hz,1h),7.91(d,j=9.0hz,1h),8.15(d,j=1.5hz,2h)。13cnmr(600mhz,cdcl3):δ19.90,31.97,34.72,109.26,114.32,116.31,116.74,122.33,123.50,123.60,123.69,125.63,127.36,138.84,140.34,141.36,143.20,145.06,149.73。c34h36n4na的计算值523.2832,实测值523.2827。对比实施例8:发光材料2-czph的合成路线如下:8a的合成:向带有磁力搅转子和冷凝管的干燥三口烧瓶中依次加入4-溴-2,6-二甲基苯甲醛(256mg,1.20mmol,1.2当量),3,6-二苯基咔唑(632mg,1.00mmol,1.0当量),pd2(dba)3(37mg,0.04mmol,8mol%),碳酸钾(415mg,3.00mmol,3当量),xantphos(16ml,0.08mmol,8mol%),然后抽换氮气三次,在氮气保护下加入甲苯(3ml)。该混合物在110℃油浴中搅拌反应26小时,薄层色谱监测至原料反应完毕,冷却至室温。加入少量的水,并用乙酸乙酯萃取。水层用乙酸乙酯萃取两次。合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚,得到中间体8a,黄色固体663mg,收率95%。2-czph的合成:在带有磁力搅拌子的反应瓶中加入中间体8a(361mg,0.80mmol,1.0当量),2-肼基吡啶(87mg,0.80mmol,1.0当量)然后抽换氮气三次,在氮气的保护下加入乙醇(4ml),在80℃的油浴锅中搅拌反应17小时。薄层色谱监测至原料反应完毕,冷却至室温。反应液移至单口瓶,真空减压蒸馏除去乙醇溶液,得到中间体8b直接进行下一步。在单口瓶中加入碘单质(244mg,0.96mmol,1.2当量),碳酸钾(332mg,2.40mmol,3.0当量)及二氯甲烷溶液(8ml),在室温下搅拌反应24小时,反应产生大量的黑色固体。反应液用硫代硫酸钠溶液洗涤,并且水相用二氯甲烷溶液萃取两遍,合并有机相,无水硫酸钠干燥,过滤。减压蒸馏除去溶剂。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/二氯甲烷/乙酸乙酯=1:2:1-1:3:1,得到2-czph,淡黄色固体367mg,两步的收率85%。1hnmr(500mhz,cdcl3):δ2.18(s,6h),6.92(td,j=6.5,1.0hz,1h),7.37(t,j=2.5hz,3h),7.49-7.52(m,6h),7.59(d,j=8.5hz,2h),7.75-7.76(m,7h),7.92(d,j=9.0hz,1h),8.15(d,j=1.5hz,2h)。13cnmr(500mhz,cdcl3):δ19.94,110.21,114.27,116.82,118.91,122.23,124.23,124.40,125.74,125.91,126.66,127.18,127.25,128.78,133.92,139.69,140.42,141.66,141.71,144.88,149.87。c38h28n4na的计算值563.2206,实测值563.2202。按照化合物1-c的合成路线,把其中的9,9-二甲基哑啶换为9,9-二苯基哑啶即得到化合物1-cph。按照化合物2-o的合成路线,把其中的吩噁嗪分别换成吩噻嗪、9,9-二甲基哑啶和9,9-二苯基哑啶即分别得到化合物2-s、2-c和2-cph。分别按照化合物1-o、1-s、1-c和1-cph的合成路线,把其中的4-溴苯甲醛换成4-溴-3,5-二甲基苯甲醛即分别得到化合物3-o、3-s、3-c和3-cph。分别按照化合物1-o、1-s、1-c和1-cph的合成路线,把其中的2-肼基吡啶换成2-肼基-4-甲基吡啶即分别得到化合物4-o、4-s、4-c和4-cph。理论计算和光物理测试数据分析说明以下所述发光材料均使用titan软件包在气相中利用lacvp**和b3lyp泛函进行理论计算所得到的结果。将本发明的1-o、1-s、1-c、3-o、3-s、3-c与对比实施例中的1-cz、1-nph2和1-czph的二面角数据进行比较,其结构式如下:表一:理论计算得到的部分化合物中受体/苯环和苯环/给体的二面角数据比较材料分子二面角(受体/苯环)二面角(苯环/给体)1-o35°83°1-s35°86°1-c36°87°3-o36°89°3-s37°88°3-c37°89°1-cz36°55°1-czph36°56°1-nph235°66°由表一可知:在受体结构一样的情况下,调控给体的结构对受体/苯环之间的二面角影响不大,其均在35°-37°之间。但是苯环/给体之间的二面角则受给体结构影响巨大,如果给体为苯噁嗪(1-o和3-o)、苯噻嗪(1-s和3-s)或哑啶(1-c和3-c)及其衍生物,由于其和苯环之间的位阻大,使苯环/给体之间的二面角高达83°-89°,使苯环/给体之间几乎处于垂直的空间结构,有效减少了苯环和给体之间的共轭,从而使其homo和lumo能够实现空间上的有效分离,如附图3所示;显然,如果在在苯环上给体的邻位引入取代基可进一步增加苯环和给体之间的位阻,进而利于homo和lumo空间上的分离,如附图4所示;而空间上分离的homo和lumo可使s1和t1之间的能级差(δest)变小。计算表明1-o、1-s、1-c、2-o、2-s、2-c、3-o、3-s和3-c的δest均在0.1ev左右,利于反系间窜越(risc),进而实现延迟荧光发光。由表一可知:相比之下,当给体为咔唑(1-cz)及其衍生物(1-czph)或二苯基胺(1-nph2)时,由于其和苯环之间的位阻减小,使苯环/给体之间的二面角大为减小(55°-66°),从而使苯环和给体之间的共轭增大,homo和lumo难以实现空间上的有效分离(附图5),δest变大,反系间窜越(risc)难以发生,难以实现延迟荧光。为了验证所设计发展材料分子的延迟荧光性质,本发明对部分材料发射光谱的溶剂效应做了进一步的测试和研究。实验选用了溶解极性依次增大的甲苯(toulene)、乙酸乙酯(ea)、四氢呋喃(thf)、二氯甲烷(dcm)和乙腈(ch3cn),如附图6、附图7、附图8和附图9所示,苯环和给体之间二面角大的1-o、1-s、1-c和2-o均显示出明显的容积效应,即随之溶剂极性的增加(附图中溶剂后括号内的数字为其介电常数,数值越大溶剂极性越大),发射光谱发生明显红移,表明由于其光激发下产生了显著的分子内电荷转移跃迁,进而证明其homo和lumo空间上的有效分离,与计算结果一致。此外,1-o、1-s、1-c和2-o在溶液中的发射光谱和最大发射波长均处于可见光区,可作为oled发光材料。作为比较,本发明亦合成了下述给体为咔唑及其衍生物的四个分子。其发射光谱的溶剂效应分别如附图10、附图11、附图12和附图13所示。从图中可知,四个分子均无明显的溶剂效应,即使对于受体和苯环之间位阻增大的2-cz、2-cztbu和2-czph亦如此,表明其在光激发下未产生显著的分子内电荷转移跃迁,进而证明其homo和lumo空间上没有有效分离,与计算结果一致。此外,1-cz、2-cz、2-cztbu和2-czph在溶液中的发射光谱和最大发射波长大部分处于紫外的不可见光区,难以作为oled发光材料使用。热活化延迟荧光发光的实现需要精巧的分子设计,一般要采用给体-受体分子体系,并且要严格调控给体和受体的分子结构,使分子的最高占有轨道(homo)和最低非占有轨道(lumo)能够实现空间上的分离。而空间上分离的homo和lumo可促进激发态分子由lumo到homo之间的电荷转移(分子内电荷转移),进而辐射发光;同时亦可使低激发单线态(s1)和最低激发三线态(t1)之间的能级差(δest)变小,只有当足够小时(≤0.2ev),才能发生快速的反系间窜越(risc),进而实现延迟荧光发光。附图14是部分材料分子掺杂的dpepo薄膜的瞬态发光光谱。depeo结构如下式所示:由附图14可知1-o、1-s和2-o均显示出两个辐射跃迁发光寿命,分别为短寿命的s1→s0跃迁和长寿命的t1→s1跃迁,证明延迟荧光的发生,与附图1一致。相比之下,2-cztbu、2-czph和2-cz均只有短寿命的s1→s0跃迁,为荧光发光,无延迟荧光。由此得到的含苯并[c][1,2,5]噻二唑衍生物结构单元发光材料光物理性质数据如下表二所示。表二:部分材料分子在室温下二氯甲烷(dcm)溶液中的光致发光量子效率从表二可知本发明申请的含吡啶并三氮唑受体结构单元的延迟荧光发光材料可强烈发光。本发明所述的吡啶并三氮唑受体结构单元的延迟荧光发光材料在作为有机电致发光器件的发光层中的应用。在有机发光元件中,从正负两电极向发光材料中注入载子,产生激发态的发光材料并使其发光。通过通式(1)代表的本发明的化合物可作为延迟荧光发光材料应用于有机光致发光元件或有机电致发光元件等优异的有机发光元件。有机光致发光元件具有在衬底上至少形成了发光层的结构。另外,有机电致发光元件具有至少形成了阳极、阴极、及阳极和阴极之间的有机层的结构。有机层至少包含发光层,可以仅由发光层构成,也可以除发光层以外具有1层以上的有机层。作为这种其它有机层,可以列举空穴传输层、空穴注入层、电子阻挡层、空穴阻挡层、电子注入层、电子传输层、激子阻挡层等。空穴传输层也可以是具有空穴注入功能的空穴注入传输层,电子传输层也可以是具有电子注入功能的电子注入传输层。具体的有机发光元件的结构示意如图15所示。在图15中,从下到上共7层,依次表示衬底、阳极、空穴注入层、空穴传输层、发光层、电子传输层和阴极,其中发光层为客体材料掺杂入主体材料的混合层。将实施例1-4中所表示的化合物作为磷光发光材料应用于oled器件,结构表示为:ito/hatcn(10nm)/tapc(65nm)/cbp:实施例1-4中所表示的化合物(10-20wt.%,20nm)/bepp2(10nm)/li2co3:bepp2(5%,30nm)/li2co3(1nm)/al(100nm)其中,ito为透明阳极;hatcn是空穴注入层,tcta是空穴传输层,cbp是主体材料,实施例1-4中所表示的化合物(10-20wt.%是掺杂浓度,20nm是发光层的厚度)是客体材料,bepp2为电子传输层,li2co3为电子注入层,al为阴极。括号中单位为纳米(nm)的数字为薄膜的厚度。需要说明的是,所述结构为本发明延迟荧光发光材料的一个应用的举例,不构成本发明所示发光材料的具体oled器件结构的限定,延迟荧光发光材料也不限于实施例1-4中所表示的化合物。器件中应用材料的分子式如下:本领域的普通技术人员可以理解,上述各实施方式是实现本发明的具体实施例,而在实际应用中,可以在形式上和细节上对其作各种改变,而不偏离本发明的精神和范围。例如,在不背离本发明的精神的情况下,这里描述的许多取代基结构可以用其它结构代替。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1