雄甾烷衍生物及其制备方法与应用与流程
本发明涉及医药
技术领域:
,尤其涉及雄甾烷衍生物及其制备方法与应用。
背景技术:
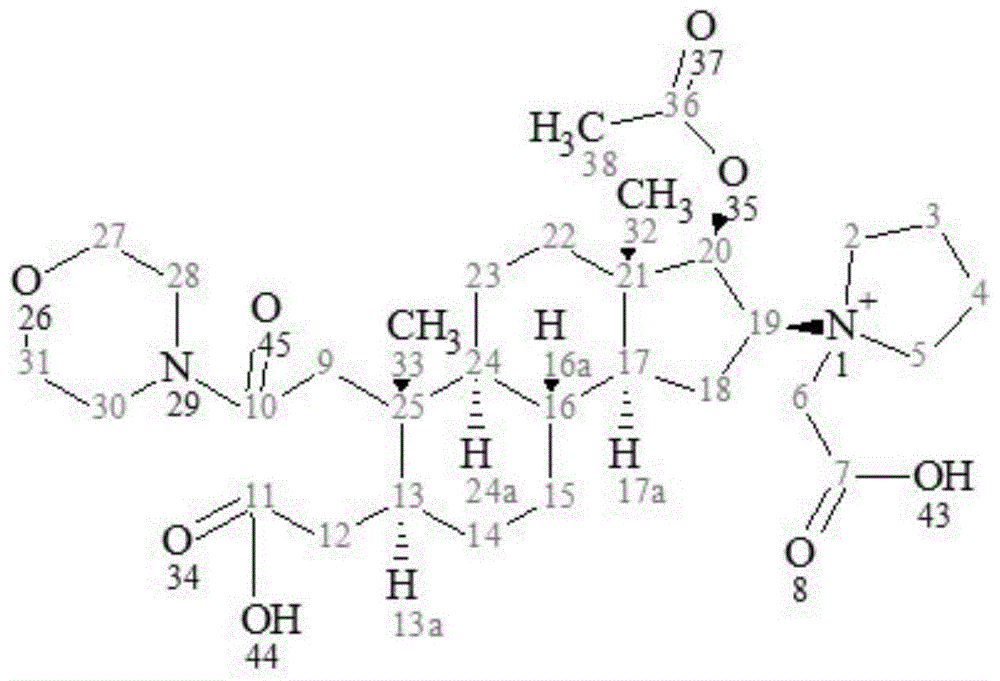
:bcr-abl融合基因,是人体细胞第9号染色体上的abl原癌基因与第24号染色体上的bcr基因相互易位形成的融合基因,可引起蛋白激酶持续性激活,使白细胞过分增殖而出现慢性髓性白血病(cml)。全球用于治疗cml的bcr-abl酪氨酸激酶抑制剂共有17款(包括上市和研发进行中药物),除了已获批上市的7款药物,在研药物包括asciminib(临床三期,诺华)、cmlvaxb2a2-25(临床二期,莱比锡大学)、pf-114(临床二期,fusionpharma)、an-019(临床二期,natco)、sun-k706(临床二期,sunpharmaadvancedresearch)、耐克替尼(临床二期,中国科学院广州生物医药与健康研究院,广州顺健生物医药科技)、dasatinibnanoparticleformulation(临床一期,xspraypharma)、etc-206(临床一期,experimentaltherapeuticscentre)、adaphostin(临床一期,mayoclinic,美国国立癌症研究所)、美迪替尼(临床一期,哈尔滨誉衡药业),提供更多的用于治疗cml的bcr-abl酪氨酸激酶抑制剂,将对改善肿瘤患者的经济负担以及提高肿瘤临床治疗效果具有重要意义。技术实现要素:本发明的目的在于提供一种雄甾烷衍生物及其制备方法与应用,本发明提供的雄甾烷衍生物对于bcr-abl酪氨酸激酶及对应mrna水平具有非常好的抑制作用,并且所述雄甾烷衍生物对于hedgehog信号通路同样具有非常好的抑制作用,在制备慢性髓性白血病药物中具有良好的应用前景。为了实现上述发明目的,本发明提供以下技术方案:本发明提供了一种雄甾烷衍生物,具有式a或式b所示结构:本发明提供了上述技术方案所述雄甾烷衍生物的制备方法,具有式a所示结构的雄甾烷衍生物的制备方法包括以下步骤:(1)将具有式ii所示结构的化合物进行氧化反应,得到具有式iii所示结构的化合物;(2)将所述具有式iii所示结构的化合物与吗啉进行缩合反应,得到具有式iv所示结构的化合物;(3)将所述具有式iv所示结构的化合物与乙酰氯进行酯化反应,得到具有式v所示结构的化合物;(4)将所述具有式v所示结构的化合物与3-溴丙酸进行季铵化反应,得到具有式a所示结构的雄甾烷衍生物;具有式b所示结构的雄甾烷衍生物的制备方法包括以下步骤:(a)将具有式ii所示结构的化合物与乙醇胺进行取代反应,得到具有式vi所示结构的化合物;(b)将所述具有式vi所示结构的化合物与甲酸甲酯进行胺酯交换反应,得到具有式vii所示结构的化合物;(c)将所述具有式vii所示结构的化合物与乙酰氯进行酯化反应,得到具有式viii所示结构的化合物;(d)将所述具有式viii所示结构的化合物与3-溴丙烯进行季铵化反应,得到具有式b所示结构的雄甾烷衍生物;其中,式ii~式viii所示结构的化合物如下所示:优选地,所述步骤(1)中氧化反应采用的氧化剂为双氧水,所述具有式ii所示结构的化合物与双氧水中h2o2的摩尔比为10∶(200~300);所述步骤(2)中具有式iii所示结构的化合物与吗啉的摩尔比为1∶(1~2);所述步骤(3)中具有式iv所示结构的化合物与乙酰氯的摩尔比为10∶(15~30);所述步骤(4)中具有式v所示结构的化合物与3-溴丙酸的摩尔比为1∶(1~2)。优选地,所述步骤(1)中氧化反应的温度为85~95℃,时间为18~22h;所述步骤(2)中缩合反应的温度为15~30℃,时间为18~20h;所述步骤(3)中酯化反应的温度为0~10℃,时间为25~35min;所述步骤(4)中季铵化反应的温度为0~10℃,时间为2~5h。优选地,所述步骤(4)中季铵化反应后还包括:在搅拌条件下,将季铵化反应后所得反应液滴加至0~10℃无水乙醚中进行析晶,之后经过滤和干燥,得到具有式a所示结构的雄甾烷衍生物。优选地,所述步骤(a)中具有式ii所示结构的化合物与乙醇胺的摩尔比为10∶(12~30);所述步骤(b)中具有式vi所示结构的化合物与甲酸甲酯的摩尔比为10∶(20~40);所述步骤(c)中具有式vii所示结构的化合物与乙酰氯的摩尔比为10∶(10~30);所述步骤(d)中具有式viii所示结构的化合物与3-溴丙烯的摩尔比为10∶(10~15)。优选地,所述步骤(a)中取代反应的温度为100~120℃,时间为30~40h;所述步骤(b)中胺酯交换反应的温度为15~30℃,时间为10~20h;所述步骤(c)中酯化反应的温度为0~10℃,时间为25~35min;所述步骤(d)中季铵化反应的温度为0~10℃,时间为2~5h。优选地,所述步骤(d)中季铵化反应后还包括:在搅拌条件下,将季铵化反应后所得反应液滴加至0~10℃无水乙醚中进行析晶,之后经过滤和干燥,得到具有式b所示结构的雄甾烷衍生物。本发明提供了上述技术方案所述雄甾烷衍生物在制备治疗慢性髓性白血病药物中的应用。优选地,所述治疗慢性髓性白血病药物包括hedgehog信号通路抑制剂药物、bcr-abl酪氨酸激酶抑制剂药物或bcr-abl酪氨酸激酶mrna抑制剂药物。本发明提供了具有式a或式b所示结构的雄甾烷衍生物。本发明提供的具有式a或式b所示结构的雄甾烷衍生物对于bcr-abl酪氨酸激酶及对应mrna水平具有非常好的抑制作用,并且所述雄甾烷衍生物对于hedgehog信号通路同样具有非常好的抑制作用,在制备慢性髓性白血病药物中具有良好的应用前景,从而为慢性髓性白血病患者提供一种有效的治疗药物。测试例1的试验结果表明,本发明提供的雄甾烷衍生物对bcr-abl酪氨酸激酶及对应mrna水平具有明显的抑制作用,可以用于bcr-abl激酶突变的慢性髓性白血病的治疗。测试例2的试验结果表明,使用小鼠[s12]和人[mz24]gli报告细胞系测得本发明提供的雄甾烷衍生物的抑制hedgehog途径信号传导的平均ic50值分别为0.019μm和0.018μm,均低于现有经fda批准的hedgehog通路抑制剂vismodegib的平均ic50值0.022μm,以及中国专利201910706129.7公开的具有式i所示结构的雄甾烷衍生物的平均ic50值0.020μm,说明本发明提供的雄甾烷衍生物对hedgehog途径信号具有非常好的抑制作用。本发明还提供了具有式a或式b所示结构的雄甾烷衍生物的制备方法,本发明提供的方法具有步骤简短、操作简单、总反应收率高、所得产品纯度较高且利于工业化生产等诸多优势。附图说明图1为实施例1制备的具有式a所示结构化合物中原子位置标识示意图;图2为实施例2制备的具有式b所示结构化合物中原子位置标识示意图。具体实施方式本发明提供了一种雄甾烷衍生物,具有式a或式b所示结构:在本发明中,所述具有式a所示结构的雄甾烷衍生物记为化合物a,根据iupac命名法,所述化合物a的化学名称为:1-((2s,3r,3as,5as,6s,7s,9ar,9bs)-3-acetoxy-7-(carboxymethyl)-3a,6-dimethyl-6-(2-morpholino-2-oxoethyl)dodecahydro-1h-cyclopenta[a]naphthalen-2-yl)-1-(carboxymethyl)pyrrolidin-1-iumbromide;所述具有式b所示结构的雄甾烷衍生物记为化合物b,根据iupac命名法,所述化合物b的化学名称为:1-((2s,3s,5s,8r,9s,10s,13s,14s,16s,17r)-17-acetoxy-3-hydroxy-2-(n-(2-hydroxyethyl)formamido)-10,13-dimethylhexadecahydro-1h-cyclopenta[a]phenanthren-16-yl)-1-allylpyrrolidin-1-iumbromide。本发明提供了上述技术方案所述雄甾烷衍生物的制备方法,以下就化合物a和化合物b的制备方法进行分别说明。在本发明中,化合物a的制备方法包括以下步骤:(1)将具有式ii所示结构的化合物进行氧化反应,得到具有式iii所示结构的化合物;(2)将所述具有式iii所示结构的化合物与吗啉进行缩合反应,得到具有式iv所示结构的化合物;(3)将所述具有式iv所示结构的化合物与乙酰氯进行酯化反应,得到具有式v所示结构的化合物;(4)将所述具有式v所示结构的化合物与3-溴丙酸进行季铵化反应,得到具有式a所示结构的雄甾烷衍生物;其中,式ii~式v所示结构的化合物如下所示:本发明将具有式ii所示结构的化合物(记为化合物ii)进行氧化反应,得到具有式iii所示结构的化合物(记为化合物iii)。本发明对所述化合物ii的来源没有特殊限定,采用本领域技术人员熟知的市售商品即可;所述化合物ii的中文名称为:(2α,3α,5α,16β,17β)-2,3-环氧-16-(1-吡咯烷基)-雄甾烷-17-醇,cas号为:119302-19-1。在本发明中,所述氧化反应采用的氧化剂优选为双氧水,所述双氧水的浓度优选为30wt%;所述氧化反应优选在催化剂存在条件下进行,所述催化剂优选为三水合磷钨酸铵;其中,所述化合物ii、双氧水中h2o2以及三水合磷钨酸铵的摩尔比优选为10∶(200~300)∶(1~3),更优选为10∶300∶1。在本发明中,所述氧化反应所用溶剂优选为水;本发明对所述溶剂的用量没有特殊要求,能够将反应物溶解即可;在本发明的实施例中,所述水与化合物ii的用量比优选为1.00l∶100g。本发明优选在搅拌条件下将化合物ii和溶剂混合,之后将所得混合物与催化剂和氧化剂混合,将体系温度升温至氧化反应的温度,进行氧化反应。在本发明中,所述氧化反应的温度优选为85~95℃,更优选为90℃;时间优选为18~22h,更优选为20h。本发明将各组分混合后,优选以15~20℃/h的速率升温至氧化反应的温度,避免因双氧水加热会分解释放出气体而引起冲料。本发明优选采用双氧水作为氧化剂,将化合物ii中的环氧基团氧化成羧酸,反应条件温和,对环境友好,对于底物的官能团耐受性好,副反应少。氧化反应后,本发明优选将所得反应液降温至室温(在本发明的实施例中,具体是指25℃),之后用乙酸乙酯萃取,所得有机层用饱和食盐水洗涤后经无水硫酸钠干燥,过滤,所得滤液浓缩后经硅胶柱层析纯化,得到淡黄色油状物,即化合物iii。将所述反应液降温至室温后,本发明优选利用淀粉碘化钾试纸测试反应液,若所述淀粉碘化钾试纸不变色,则进行后续处理;若所述淀粉碘化钾试纸变色,优选向所述反应液中加入饱和的硫代硫酸钠,至淀粉碘化钾试纸不变色。在本发明中,纯化所用试剂优选为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比优选为5∶1。得到化合物iii后,本发明将所述化合物iii与吗啉进行缩合反应,得到具有式iv所示结构的化合物(记为化合物iv)。在本发明中,所述化合物iii与吗啉的摩尔比优选为1∶(1~2),更优选为1∶1。在本发明中,所述缩合反应优选在有机溶剂、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)和三乙胺存在条件下进行;所述化合物iii、hatu和三乙胺的摩尔比为10∶(12~20)∶(20~30),更优选为10∶12∶20。在本发明中,所述有机溶剂优选为二氯甲烷(dcm),本发明对所述有机溶剂的用量没有特殊要求,能够将反应物溶解即可;在本发明的实施例中,所述有机溶剂与化合物iii的用量比优选为453ml∶111.1mmol。本发明优选在搅拌条件下将有机溶剂与化合物iii混合,在氮气保护条件下,将体系的温度降到0~10℃(进一步优选为0℃),加入三乙胺(et3n)和hatu,搅拌10~20min,加入吗啉,搅拌8~12min,然后将体系温度升温至缩合反应的温度,进行缩合反应。在本发明中,所述缩合反应的温度优选为15~30℃,更优选为20~25℃;在本发明的实施例中,具体是在室温(25℃)条件下进行所述缩合反应,即不需要额外的加热或降温;所述缩合反应的时间优选为18~20h,更优选为20h。本发明将各组分混合并搅拌8~12min后,优选以10~15℃/h的速率升温至缩合反应的温度,有利于保证反应顺利进行。本发明优选通过hatu实现化合物iii中羧基的活化,然后再加入吗啉进行缩合反应,反应条件温和,选择性好,且反应活性高。缩合反应后,本发明优选将所得反应液用氢氧化钠水溶液(浓度优选为1mol/l)萃取,所得水相用二氯甲烷继续萃取,之后将所得水相用盐酸调节ph值至5~6,再用二氯甲烷进行萃取,将所得有机相用饱和食盐水洗涤后经无水硫酸钠干燥,过滤,所得滤液浓缩后经硅胶柱层析纯化,得到白色固体,即化合物iv;在本发明中,纯化所用试剂优选为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比优选为5∶1。得到化合物iv后,本发明将所述化合物iv与乙酰氯进行酯化反应,得到具有式v所示结构的化合物(记为化合物v)。在本发明中,所述化合物iv与乙酰氯的摩尔比优选为10∶(15~30),更优选为10∶30。在本发明中,所述酯化反应优选在有机溶剂和三乙胺存在条件下进行,所述化合物iv和三乙胺的摩尔比为10∶(15~40),更优选为10∶40。在本发明中,所述有机溶剂优选为二氯甲烷,本发明对所述有机溶剂的用量没有特殊要求,能够将反应物溶解即可;在本发明的实施例中,所述化合物iv和有机溶剂的用量比优选为46.2mmol∶220ml。本发明优选在搅拌条件下将有机溶剂和化合物iv混合,在氮气保护条件下,将所得体系的温度降到0~10℃,加入et3n和乙酰氯,进行酯化反应。在本发明中,所述酯化反应的温度优选为0~10℃,更优选为0℃;时间优选为25~35min,更优选为30min。本发明的酯化反应中优选以三乙胺作为碱,化合物iv与乙酰氯经酯化反应,得到化合物v,反应条件温和。所述酯化反应后,本发明优选将所得反应液用水萃取,所得水相再用二氯甲烷萃取,将所得有机相用饱和食盐水洗涤后经无水硫酸钠干燥,过滤,所得滤液浓缩后,再经硅胶柱层析纯化,得到白色固体,即化合物v;在本发明中,纯化所用试剂优选为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比优选为5∶1。得到化合物v后,本发明将所述化合物v与3-溴丙酸进行季铵化反应,得到化合物a。在本发明中,所述化合物v与3-溴丙酸的摩尔比优选为1∶(1~2),更优选为1∶1.2。在本发明中,所述季铵化反应优选在有机溶剂存在条件下进行,所述有机溶剂优选为二氯甲烷,本发明对所述有机溶剂的用量没有特殊要求,能够将反应物溶解即可;在本发明的实施例中,所述化合物v和有机溶剂的用量比优选为0.0204mol∶106ml。本发明优选在0~10℃、搅拌条件下,向二氯甲烷中依次加入化合物v和3-溴丙酸,然后进行季铵化反应。在本发明中,所述季铵化反应的温度优选为0~10℃,更优选为10℃;时间优选为2~5h,更优选为3h。所述季铵化反应后,本发明优选在搅拌条件下,将所得反应液滴加至0~10℃(优选为5℃)的无水乙醚中进行析晶,具体是滴毕后保温搅拌10~30min,之后过滤,将所得滤饼常温减压干燥,得到白色固体,即为化合物a。本发明优选利用化合物a在无水乙醚中几乎不溶的特点,将反应液加至无水乙醚中析出产品,同时达到纯化的目的,该后处理方法简单易操作且产品收率高。在本发明中,化合物b的制备方法包括以下步骤:(a)将具有式ii所示结构的化合物与乙醇胺进行取代反应,得到具有式vi所示结构的化合物;(b)将所述具有式vi所示结构的化合物与甲酸甲酯进行胺酯交换反应,得到具有式vii所示结构的化合物;(c)将所述具有式vii所示结构的化合物与乙酰氯进行酯化反应,得到具有式viii所示结构的化合物;(d)将所述具有式viii所示结构的化合物与3-溴丙烯进行季铵化反应,得到具有式b所示结构的雄甾烷衍生物;其中,式ii所示结构的化合物与上述技术方案一致,式vi~式viii所示结构的化合物如下所示:本发明将具有式ii所示结构的化合物与乙醇胺进行取代反应,得到具有式vi所示结构的化合物(记为化合物vi)。在本发明中,制备化合物b时所需起始原料为具有式ii所示结构的化合物(即化合物ii),与上述制备化合物a时的起始原料相同。在本发明中,所述化合物ii与乙醇胺的摩尔比优选为10∶(12~30),更优选为10∶15。在本发明中,所述取代反应优选在有机溶剂存在条件下进行,所述有机溶剂优选为正丁醇,本发明对所述有机溶剂的用量没有特殊要求,能够将反应物溶解即可;在本发明的实施例中,所述化合物ii和有机溶剂的用量比优选为278mmol∶500ml。本发明优选将化合物ii与正丁醇混合,之后在20~25℃、搅拌条件下向所得体系中加入乙醇胺进行取代反应。在本发明中,所述取代反应的温度优选为100~120℃,更优选为120℃;时间优选为30~40h,更优选为30h。本发明采用乙醇胺作为亲核试剂,反应条件温和,产率高,对于底物的官能团耐受性好,副反应少。所述取代反应后,本发明优选将所得反应液降温到室温,之后与水混合,用甲基叔丁基醚萃取所得混合物,采用饱和食盐水洗涤所得有机层后经无水硫酸钠干燥,过滤,所得滤液浓缩后经硅胶柱层析纯化,得到白色固体,记为化合物vi;在本发明中,纯化所用试剂优选为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比优选为5∶1。得到化合物vi后,本发明将所述化合物vi与甲酸甲酯进行胺酯交换反应,得到具有式vii所示结构的化合物(记为化合物vii)。在本发明中,所述胺酯交换反应优选在甲醇和甲醇钠存在条件下进行;所述化合物vi、甲醇钠和甲酸甲酯的摩尔比优选为10∶(10~40)∶(20~40),更优选为10∶40∶40;本发明对所述甲醇的用量没有特殊要求,其作为有机溶剂能够将反应物溶解即可;在本发明的实施例中,所述化合物vi和甲醇的用量比优选为252mmol:1l。本发明优选在搅拌条件下将甲醇与化合物vi混合,将所得体系的温度降到0℃,加入甲醇钠和甲酸甲酯,之后将所得体系自然升温到胺酯交换反应的温度,进行胺酯交换反应;在本发明中,所述胺酯交换反应的温度优选为15~30℃,更优选为20~25℃;在本发明的实施例中,具体是在室温(25℃)条件下进行所述胺酯交换反应,即不需要额外的加热或降温;所述胺酯交换反应的反应时间优选为10~20h,更优选为10h。本发明的胺酯交换反应中优选以甲醇钠作为碱,甲酸甲酯作为交换试剂,与化合物vi结构中的仲胺进行酯交换反应,得到化合物vii;本发明的方法反应条件温和,选择性好,且产物收率高。所述胺酯交换反应后,本发明优选将所得反应液浓缩至干,所得残余固体用水溶解,之后用甲基叔丁基醚萃取,采用盐酸将所得水相的ph值调节至6~7,然后用乙酸乙酯萃取,所得有机相用饱和食盐水洗涤后经无水硫酸钠干燥,过滤,所得滤液浓缩后得黄色固体,之后经硅胶柱层析纯化,得到白色固体,即为化合物vii;在本发明中,纯化所用试剂优选为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比优选为5∶1。得到化合物vii后,本发明将所述化合物vii与乙酰氯进行酯化反应,得到具有式viii所示结构的化合物(记为化合物viii)。在本发明中,所述化合物vii与乙酰氯的摩尔比优选为10∶(10~30),更优选为10∶15。在本发明中,所述酯化反应优选在有机溶剂和三乙胺存在条件下进行,所述化合物vii和三乙胺的摩尔比为10∶(15~40),更优选为10∶30。在本发明中,所述有机溶剂优选为二氯甲烷,本发明对所述有机溶剂的用量没有特殊要求,能够将反应物溶解即可;在本发明的实施例中,所述化合物vii和有机溶剂的用量比优选为207mmol∶1l。本发明优选在搅拌条件下将有机溶剂和化合物vii混合,在氮气保护条件下,将所得体系的温度降到0~10℃,加入et3n和乙酰氯,进行酯化反应。在本发明中,所述酯化反应的温度优选为0~10℃,更优选为5℃;时间优选为25~35min,更优选为30min。本发明的酯化反应中优选以三乙胺作为碱,化合物vii与乙酰氯经酯化反应,得到化合物viii,反应条件温和。所述酯化反应后,本发明优选将所得反应液用水萃取,所得水相再用二氯甲烷萃取,将所得有机相用饱和食盐水洗涤后经无水硫酸钠干燥,过滤,所得滤液浓缩后,再经硅胶柱层析纯化,得到白色固体,即化合物viii;在本发明中,纯化所用试剂优选为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比优选为5∶1。得到化合物viii后,本发明将所述化合物viii与3-溴丙烯进行季铵化反应,得到具有式b所示结构的雄甾烷衍生物(即化合物b)。在本发明中,所述化合物viii与3-溴丙烯的摩尔比优选为10∶(10~15),更优选为10∶12。在本发明中,所述季铵化反应优选在有机溶剂存在条件下进行,所述有机溶剂优选为二氯甲烷,本发明对所述有机溶剂的用量没有特殊要求,能够将反应物溶解即可;在本发明的实施例中,所述化合物viii和有机溶剂的用量比优选为89mmol∶400ml。本发明优选在0~10℃、搅拌条件下,将化合物viii、3-溴丙烯和有机溶剂混合进行季铵化反应;在本发明中,所述季铵化反应的温度优选为0~10℃,更优选为10℃;时间优选为2~5h,更优选为3h。所述季铵化反应后,本发明优选在搅拌条件下,将所得反应液滴加至0~10℃无水乙醚中进行析晶,具体是滴毕后保温搅拌10~30min,之后过滤,所得滤饼常温减压干燥后得到白色固体,即为化合物b。本发明优选利用化合物b在无水乙醚中几乎不溶的特点,将反应液加至无水乙醚中析出产品,同时达到纯化的目的,该后处理方法简单易操作且产品收率高。本发明提供了上述技术方案所述雄甾烷衍生物在制备治疗慢性髓性白血病药物中的应用。在本发明中,所述治疗慢性髓性白血病药物优选包括hedgehog信号通路抑制剂药物、bcr-abl酪氨酸激酶抑制剂药物或bcr-abl酪氨酸激酶mrna抑制剂药物。在本发明中,所述治疗慢性髓性白血病药物优选包括活性组分与载体;所述活性组分为化合物a或化合物b;所述载体为药学可接受的载体,包括但不限于以下物质中的一种或多种:甘露醇,山梨醇,焦亚硫酸钠,亚硫酸氢钠,硫代硫酸钠,盐酸半胱氨酸,巯基乙酸,蛋氨酸,维生素c,edta二钠,edta钙钠,一价碱金属的碳酸盐、醋酸盐、磷酸盐或其水溶液,盐酸,醋酸,硫酸,磷酸,氨基酸,氯化钠,氯化钾,乳酸钠,木糖醇,麦芽糖,葡萄糖,果糖,右旋糖苷,甘氨酸,淀粉,蔗糖,乳糖,甘露糖醇,硅衍生物,纤维素或其衍生物,藻酸盐,明胶,聚乙烯吡咯烷酮,甘油,土温80,琼脂,碳酸钙,碳酸氢钙,表面活性剂,聚乙二醇,环糊精,β-环糊精,磷脂类材料,高岭土,滑石粉,硬脂酸钙,硬脂酸镁。在本发明中,所述药物中活性组分的含量优选为0.1~99.9wt.%。本发明对所述药物的剂型没有特殊要求,具体可以为片剂、胶囊剂、口服液、口含剂、颗粒剂、冲剂、丸剂、散剂、膏剂、丹剂、混悬剂、粉剂、溶液剂、注射剂、栓剂、酊剂、膏剂、霜剂、喷雾剂、滴剂或贴剂;优选的是片剂、散剂、颗粒剂、酊剂、丸剂、胶囊剂、口服液、雾化吸入剂或注射剂。本发明可根据要制备的药物剂型添加其他辅料。本发明对所述辅料的种类没有特殊要求,采用本领域熟知的辅料即可。在本发明中,当所述药物的剂型为口服给药的制剂时,所述药物优选还包括赋形剂,本发明对所述赋形剂的种类没有特殊要求,采用本领域熟知的赋形剂即可;具体可以为粘合剂、填充剂、稀释剂、压片剂、润滑剂、崩解剂、着色剂、调味剂或湿润剂。在本发明中,所述填充剂优选包括纤维素、甘露糖醇、乳糖和其它类似的填充剂中的一种或多种,本发明对所述其它类似的填充剂的具体种类没有特殊要求,本领域熟知的具有类似功能的填充剂均可;所述崩解剂优选包括淀粉、聚乙烯吡咯烷酮和淀粉衍生物中的一种或多种,本发明对所述淀粉衍生物的具体种类没有特殊要求,本领域熟知的淀粉衍生物均可,例如羟基乙酸淀粉钠;所述润滑剂优选包括硬脂酸镁;所述湿润剂优选包括十二烷基硫酸钠。下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例1按以下反应流程制备具有式a所示结构的雄甾烷衍生物:(1)在搅拌条件下,向三口反应瓶中依次加入水(1.00l)和具有式ii所示结构的化合物(100g,278mmol),之后加入三水合磷钨酸铵(83.0g,27.8mmol)和浓度为30wt%的双氧水(945g,8.34mol),以18℃/h的速率升温到90℃,反应20h;反应结束后,将所得反应液降温到室温(25℃,利用淀粉碘化钾试纸测试反应液,所述淀粉碘化钾试纸不变色),采用乙酸乙酯萃取3次,每次萃取所用乙酸乙酯的体积为500ml,合并有机层,并用食盐水洗涤,洗涤后所得有机相用无水硫酸钠干燥,过滤,所得滤液浓缩后经硅胶柱层析纯化(纯化试剂为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比为5∶1),得到淡黄色油状物45.3g,收率为40.0%,经lc-ms方法(检测条件见表1)检测,纯度为95.6%。对所述淡黄色油状物进行表征,表征数据具体如下:ms:m/z:408[m+h]+。以上结果说明所述淡黄色油状物即为具有式iii所示结构的化合物。表1lc-ms方法检测条件(2)在搅拌条件下,向三口瓶中加入二氯甲烷(dcm,453ml)和具有式iii所示结构的化合物(45.3g,111.1mmol),在氮气保护条件下,将所得体系的温度降到0℃,加入三乙胺(et3n,22.5g,222.2mmol)和2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu,50.7g,133.4mmol),搅拌15min,加入吗啉(10.7g,111.1mmol),搅拌10min,以12℃/h的速率升温到室温,继续搅拌反应20h;反应结束后,将所得反应液倒入500ml浓度为1mol/l的氢氧化钠水溶液中进行萃取,水相用二氯甲烷继续萃取2次,每次萃取所用二氯甲烷的体积为200ml,收集水相,将所述水相用2mol/l的盐酸调节ph值至5~6,之后用二氯甲烷进行萃取3次,每次萃取所用二氯甲烷的体积为500ml,合并有机相,将所述有机相用饱和食盐水洗涤后经无水硫酸钠干燥,过滤,所得滤液浓缩后得到黄色固体,经硅胶柱层析纯化(纯化试剂为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比为5∶1),得到白色固体22.0g,收率为42.0%,纯度为90.1%。对所述白色固体进行表征,表征数据具体如下:ms:m/z:477[m+h]+。以上结果说明所述白色固体即为具有式iv所示结构的化合物。(3)在搅拌条件下,向三口瓶中加入dcm(220ml)和具有式iv所示结构的化合物(22.0g,46.2mmol),在氮气保护条件下,将所得体系的温度降到0℃,加入et3n(18.7g,184.8mmol)和乙酰氯(10.9g,138.6mmol),在0℃、搅拌条件下反应30min;反应结束后,将所得反应液倒入500ml的水中进行萃取,所得水相用二氯甲烷继续萃取2次,每次萃取所用二氯甲烷的体积为200ml,合并有机相,将所述有机相用饱和食盐水洗涤后经无水硫酸钠干燥,过滤,所得滤液浓缩后得黄色固体,经硅胶柱层析纯化(纯化试剂为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比为5∶1),得到白色固体10.6g,收率为65.0%,纯度为96.1%。对所述白色固体进行表征,表征数据具体如下:ms:m/z:519[m+h]+。以上结果说明所述白色固体即为具有式v所示结构的化合物。(4)在10℃、搅拌条件下,向106ml二氯甲烷中依次加入具有式v所示结构的化合物(10.6g,0.0204mol)和3-溴丙酸(3.4g,0.0245mol),10℃条件下搅拌反应3h后,保持搅拌条件,将所得反应液滴加至5℃无水乙醚中(1.0kg),滴毕后5℃条件下搅拌10min,过滤,将所得滤饼常温减压干燥后得到白色固体8.0g,收率为60.0%,经lc-ms方法检测(检测条件见表1),纯度为96%以上。对所述白色固体进行表征,表征数据具体如下:高分辨质谱:高分辨质谱:m/z577.3470m+;模拟分子式:c31h49n2o8+;理论计算值:577.3489;不饱和度:9;核磁共振:1h-nmr(400mhz,cd3cn∶d2o=1∶1)显示脂肪区信号重叠,通过hsqc可以确定存在3个甲基信号δh1.149,δh1.175,δh2.546;存在16个亚甲基信号,分别为δh1.787(2h),δh2.516(4h),δh1.949和δh1.646,δh2.094和δh2.492,δh1.317和δh2.036,δh2.319和δh2.816,δh2.703(2h),δh1.724和δh2.147,δh3.851(2h),δh3.888(2h),δh4.522(2h),δh4.039和δh4.248,δh4.088和δh4.224,δh3.972(2h)和δh3.989(2h),其中δh3.851(2h),δh3.888(2h),δh4.522(2h),δh4.039和δh4.248,δh4.088和δh4.224,δh3.972(2h)和δh3.989(2h)均与电负性较强原子相连;存在6个次甲基信号δh1.881,δh2.749,δh1.489,δh2.119,δh5.000,δh5.472,其中δh5.000,δh5.472与电负性较强的原子相连。13c-nmr(100mhz,cd3cn∶d2o=1∶1)显示存在4个羰基碳信号δc166.804,δc170.354,δc170.763,δc176.646;存在2个季碳信号δc39.365和δc44.381;存在6个次甲基碳信号δc33.008,δc38.585,δc46.335,δc46.488,δc69.388,δc77.743,其中δc77.743与氧原子相连,δc69.388信号很弱,出峰特别低,并且峰形宽,表明它受到氮正离子的影响;存在16个亚甲基碳信号δc21.037,δc23.107(2),δc27.001,δc27.161,δc30.456,δc35.756,δc35.851,δc37.047,δc41.202,δc45.759,δc60.515和δc63.249,δc65.035,δc65.852,δc66.019,δc60.515和δc63.249信号很弱,出峰特别低,并且峰形宽,表明它受到氮正离子的影响;存在3个甲基碳信号δc12.099,δc14.803,δc20.016,其中δc20.016为羰基邻位。通过分子式c31h49n2o8+和相对分子量577确定化合物有4个羰基,为4个不饱和度,剩余5个不饱和度推测存在5个环状结构。图1为实施例1制备的具有式a所示结构化合物中原子位置标识示意图。1h1h-cosy和tocsy中δh5.472(h-20),δh5.000(h-19),δh2.492(18),δh2.094(18),δh1.489(17),δh2.119(16a),δh1.724(22),δh2.147(22),δh1.787(23),δh1.949(15),δh2.749(24),δh2.816(12),δh2.319(12),δh1.317(14)有相关,hmbc中δh5.472(h-20)与δc12.099(c-32),δc27.161(c-18),δc37.047(c-22),δc44.381(c-21),δc170.354(c-36)有远程相关,结合剩余不饱和度,综合推测式a的母核为甾族类化合物,hmbc中δh1.149(h-32)与δc77.743(c-20),δc44.381(c-21),δc37.047(c-22)有远程相关,δh1.175(h-33)与δc39.365(25)有远程相关,证明2个甲基分别属于母核的21位和25位,符合甾族类化合物的结构特征。1h1h-cosy中δh3.989(h-31)与δh3.851相关,hmbc中δh3.989(h-31)与δc65.852(c-27)和δc45.759(c-28)有远程相关,27位亚甲基和31位亚甲基的碳氢数据显示都与氧原子相连,而且28位亚甲基和30位亚甲基的碳氢数据显示都与氮原子相连,推断存在吗啉环。hmbc中δh3.851(h-30)和δh3.888(h-28)均与δc170.763(c-10)有远程相关,δh2.703(h-9)与δc170.763(c-10),δc39.365(c-25),δc14.803(c-33)有远程相关,9位的亚甲基碳氢数据显示为酰胺羰基邻位,推断存在2-吗啉代-2-氧乙基结构片段位于25位。hmbc中δh2.546(h-38)与δc170.354(c-36)有远程相关,证明存在乙酰基,20位的次甲基碳氢数据显示与氧原子相连,hmbc中δh5.472(h-20)与δc170.354(c-36)有远程相关,推断乙酰氧基位于20位。hmbc中δh2.319(h-12)与δc176.646(c-11)有远程相关,证明存在羧甲基结构片段。hmbc中δh2.319(h-12)与δc54.428(c-24)有远程相关,表明羧甲基位于13位。hmbc中δh4.522(h-6)和δc166.804(c-7)相关,证明存在羧甲基结构片段;1h1h-cosy和tocsy中δh2.516(h-3&4)和δh4.088(h-2),δh4.248(h-5)相关,2/5位的2个亚甲基碳氢数据显示与氮原子相连,结合剩余的不饱和度,推测存在吡咯烷基;19位次甲基和6位亚甲基的碳氢数据显示都与氮原子相连,综合推测存在1-丙烯基吡咯烷鎓基位于19位。通过1h-nmr、13c-nmr、1h1h-cosy、hsqc、hmbc和tocsy表征对式a所示结构进行了碳氢数据归属。由上述表征结果可知,实施例1制备得到的白色固体即为具有式a所示结构的雄甾烷衍生物。实施例2按以下反应流程制备具有式b所示结构的雄甾烷衍生物:(1)在搅拌条件下,向三口反应瓶中依次加入正丁醇(500ml)和具有式ii所示结构的化合物(100g,278mmol),之后在温度25℃、搅拌条件下加入乙醇胺(25.6g,420mmol),升温到120℃反应30h;反应结束后,将所得反应液降温到室温,之后倒入3l水中,用甲基叔丁基醚萃取3次,每次萃取所用甲基叔丁基醚的体积为500ml,合并有机层,并用食盐水洗涤,洗涤后所得有机相以无水硫酸钠干燥,过滤,浓缩后经硅胶柱层析纯化(纯化试剂为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比为5∶1),得到白色固体106g,收率为90%,纯度为96.0%(纯度检测方法同实施例1)。对所述白色固体进行表征,表征数据具体如下:ms:m/z:421[m+h]+。以上结果说明所述白色固体即为vi所示结构的化合物。(2)在搅拌条件下,向三口瓶中加入meoh(1.00l)和具有式vi所示结构的化合物(106g,252mmol),将所得体系的温度降到0℃,加入甲醇钠(54.4g,1.00mol)和甲酸甲酯(60.53g,1.00mol),之后将所得反应液自然升温到室温,继续搅拌反应10h;反应结束后,将所得反应液浓缩至干,残余固体加1.00l的水溶解,之后用甲基叔丁基醚萃取3次,每次萃取所用甲基叔丁基醚的体积为300ml,收集水相,所述水相用2mol/l的盐酸调节ph至6~7,然后用乙酸乙酯萃取3次,每次萃取所用乙酸乙酯的体积为400ml,有机相用饱和食盐水洗涤后经无水硫酸钠干燥,过滤,所得滤液浓缩后得黄色固体,之后经硅胶柱层析纯化(纯化试剂为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比为5∶1),得到白色固体92.7g,收率为82.0%,纯度为96.1%。对所述白色固体进行表征,表征数据具体如下:ms:m/z:449[m+h]+。以上结果说明所述白色固体即为vii所示结构的化合物。(3)在搅拌条件下,向三口瓶中加入dcm(1.00l)和具有式vii所示结构的化合物(92.7g,207mmol),在氮气保护条件下,将所得体系的温度降到5℃,加入et3n(62.8g,621mmol)和乙酰氯(24.3g,310mmol),搅拌反应30min;反应结束后,将所得反应液倒入500ml的水中,进行萃取,水相再用二氯甲烷萃取2次,每次萃取所用二氯甲烷的体积为200ml,合并有机相,所述有机相用饱和食盐水洗涤后经无水硫酸钠干燥,过滤,滤液浓缩后得到黄色固体,之后经硅胶柱层析纯化(纯化试剂为二氯甲烷和甲醇,所述二氯甲烷和甲醇的体积比为5∶1),得到白色固体43.7g,收率为43%,纯度为97.0%。对所述白色固体进行表征,表征数据具体如下:ms:m/z:491[m+h]+。以上结果说明所述白色固体即为viii所示结构的化合物。(4)在10℃、搅拌条件下,向三口瓶中加入dcm(400ml)、具有式viii所示结构的化合物(43.7g,89mmol)以及3-溴丙烯(12.9g,107mol),10℃条件下搅拌反应3h;反应结束后,保持搅拌条件,将所得反应液滴加至5℃无水乙醚中(3.00kg),滴毕后5℃条件下搅拌10min,过滤,所得滤饼常温减压干燥后得到白色固体50.0g,收率为90%,纯度为96%以上(纯度检测方法同实施例1);对所述白色固体进行表征,表征数据具体如下:高分辨质谱:高分辨质谱:m/z531.3789m+;模拟分子式:c31h51n2o5+;理论计算值:531.3798;不饱和度:8;核磁共振:1h-nmr(600mhz,cd3cn∶d2o=1∶1)显示脂肪区信号重叠,通过hsqc可以确定存在3个甲基信号δh0.731,δh0.819,δh2.121;存在15个亚甲基信号,分别为δh1.426和δh1.278,δh2.060(4h),δh2.036和δh1.686,δh1.105和δh1.402,δh0.865和δh1.622,δh1.808和δh1.442,δh1.250和δh1.700,δh1.610和δh1.366,δh3.224和δh3.269,δh3.525和δh3.709,δh3.568(4h),δh3.850(2h),δh5.604和δh5.540,其中δh5.604和δh5.540为末端烯氢特征信号,δh3.224和δh3.269,δh3.525和δh3.709,δh3.568(4h),δh3.850(2h)与电负性较强原子相连;存在10个次甲基信号δh1.402,δh1.585,δh0.991,δh0.761,δh3.593,δh3.925,δh3.975,δh5.024,δh5.970,δh7.964,其中δh7.964为甲酰基质子信号,δh5.970为烯氢信号,δh3.593,δh3.925,δh3.975,δh5.024与电负性较强的原子相连。13c-nmr(150mhz,cd3cn∶d2o=1∶1)显示存在2个羰基碳信号δc165.426,δc170.304;存在2个季碳信号δc35.004和δc44.731;存在10个次甲基碳信号δc33.064,δc37.906,δc46.065,δc54.428,δc60.232,δc64.199,δc67.560,δc77.586,δc125.031,δc165.426,其中δc165.426为甲酰基碳信号,δc77.586与氧原子相连,δc125.031为烯碳信号,δc67.560信号很弱,出峰特别低,并且峰形宽,表明它受到氮正离子的影响;存在15个亚甲基碳信号δc20.158,δc22.980(2),δc26.918,δc27.136,δc30.418,δc34.603,δc37.089,δc40.451,δc42.704,δc58.242,δc62.631(2),δc64.695,δc127.664,其中δc127.664为末端烯碳信号;存在3个甲基碳信号δc12.211,δc16.053,δc20.020,其中δc20.020为羰基邻位。通过分子式c31h51n2o5+和相对分子量531确定化合物有2个羰基,1个烯烃双键,为3个不饱和度,剩余5个不饱和度推测存在5个环状结构。图2为实施例2制备的具有式b所示结构化合物中原子位置标识示意图。1h1h-cosy和tocsy中δh5.024(h-20),δh3.975(h-19),δh1.686(h-18),δh2.036(h-18),δh0.991(h-17),δh1.402(h-16),δh0.865(h-15),δh1.105(h-14),δh0.761(h-24),δh1.278(h-23),δh1.700(h-22),δh1.585(h-13),δh1.610(h-9),δh1.366(h-9),δh3.593(h-10),δh3.925(h-11),δh1.808(h-12)有相关;hmbc中δh0.991(h-17)与δc26.918(c-18),δc44.731(c-21)有远程相关,δh1.426(h-23)与δc35.004(c-25),δc54.428(c-24),δc37.906(c-13),δc44.731(c-21)有远程相关,结合剩余不饱和度,综合推测式i的母核为甾族化合物,hmbc中δh0.731(h-30)与δc44.731(c-21),δc37.089(c-22),δc46.065(c-17),δc77.586(c-20)有远程相关,δh0.819(h-31)与δc35.004(c-25),δc40.451(c-9),δc37.906(c-13),δc54.428(c-24)有远程相关,证明2个甲基分别属于母核的21位和25位,符合甾族化合物的结构特征。1h1h-cosy中h-27δh3.525/3.709与h-28δh3.224/3.269相关,2个相连的亚甲基碳氢数据显示,分别与氧原子和氮原子相连;hmbc中δh7.964(h-44)与δc60.232(c-10)有远程相关,δh3.593(h-10)与δc165.426(c-44)有远程相关,δh3.224/3.269(h-28)与δc58.242(c-27)和δc165.426(c-44)有远程相关,推断存在n-(2-羟乙基)甲酰胺基结构片段位于10位。hmbc中δh2.121(h-36)与δc170.304(c-34)有远程相关,证明存在乙酰基,20位的次甲基碳氢数据显示与氧原子相连,hmbc中δh5.024(h-20)与δc170.304(c-34)有远程相关,推断乙酰氧基位于20位。1h1h-cosy和tocsy中δh5.970(h-7)和δh3.850(h-6),δh5.604(h-8)有相关,证明存在烯丙基;1h1h-cosy和tocsy中δh3.568(h-2&5)和δh2.060(h-3&4)相关,2/5位的2个亚甲基碳氢数据显示与氮原子相连,结合剩余的不饱和度,推测存在吡咯烷基;19位次甲基和6位亚甲基的碳氢数据显示都与氮原子相连,综合推测存在1-丙烯基吡咯烷鎓基位于19位;通过1h-nmr、13c-nmr、1h1h-cosy、hsqc、hmbc和tocsy表征对式b所示结构进行了碳氢数据归属。由上述表征结果可知,实施例2制备得到的白色固体即为具有式b所示结构的雄甾烷衍生物。测试例1测试式a和式b所示结构的雄甾烷衍生物对bcr-abl酪氨酸激酶及对应mrna水平的抑制作用,并与发明专利201910706129.7中式i所示结构的化合物进行比较,具体如下:试验中使用k562细胞系(人慢性骨髓性白血病细胞系)。将k562细胞常规培养于rpmi1640培养基(含10%胎牛血清、100μg/ml青霉素和100μg/ml链霉素)中,并置于37℃、5%co2的恒温培养箱中培养。细胞隔日换液传代1次,取处于对数生长期的k562细胞进行实验。mtt法测定化合物对细胞增殖的抑制效果:将k562细胞分别接种于96孔板,每孔1×105个细胞,体积为100μl,置于37℃、5%co2恒温培养箱中培养3日。培养结束后,各组分别加入100μl用rpmi1640培养液稀释的浓度为1μmol/l、10μmol/l的式i、式a以及式b所示结构的化合物溶液,空白孔用不含有细胞的培养液代替。分别培养24h、48h、72h后向每孔加入20μlmtt溶液(5mg/ml),继续培养4h后离心,在避光的条件下小心弃去上清,然后每孔中加入150μldmso使孔内结晶充分溶解。酶标仪上读取波长490nm、参比波长620nm处各孔的吸光度(a)值。计算细胞增殖抑制率,试验重复三次。细胞增殖抑制率=(对照组a值-试验组a值)/对照组a值×100%。实时荧光定量pcr技术检测bcr/ablmrna的表达水平:收集式a以及式b所示结构的化合物处理48h后的k562细胞、trizol试剂抽提细胞的总rna进行pcr检测。以人gapdh为内参,从ncbi数据库查找bcr/abl基因dna序列,primerpremier5.0软件设计引物。gapdh上游引物:5’-ctctgctcctcctgttcgac-3’;下游引物:5’-cactgtgttggcgtacagg-3’;bcr/abl上游引物:5’-aatgccgctgagtatctgct-3’;下游引物:5’-gccatcagaagcagtattga-3’。pcr扩增条件为:94℃预变性10min,60℃退火60s,72℃延伸30s,共45个循环。实验结果利用电脑软件分析阈值循环数(ct),每个样本设3个复孔,计算取平均值。将gapdh的平均值记为ct0,实验组的平均值记为ct(x)。计算相对于gapdh的表达量,公式:2–δδct=2-[δct(x)-(ct(x)-ct0)]。具体试验结果见表2~3。表2不同浓度的受试物对k562细胞增殖的抑制率(%)根据表2中的数据可知,选择不同浓度的式i、式a和式b(0、1、10μmol/l)处理k562细胞24h、48h、72h后,采用mtt法检测其对k562细胞株的增殖抑制作用。结果表明,具有式i所示结构的化合物不能抑制k562细胞增殖,而具有式a、b所示结构的化合物对k562细胞的增殖有明显抑制作用,且抑制作用随着时间的延长而增加,具有一定的剂量依赖性。表3化合物作用于k562细胞48hbcr-abl基因的相对表达量(x±s)根据表3中的数据可知,选择不同浓度的式i、式a和式b(0、1、10μmol/l)处理k562细胞48h后,实时荧光定量法检测bcr/ablmrna的表达水平,结果表明,具有式i所示结构的化合物在0μmol/l、1μmol/l或10μmol/l浓度下均不能抑制bcr-ablmrna的表达,而具有式a、b所示结构的化合物对bcr-ablmrna具有抑制作用;此外与空白组相比,经式a和式b所示结构化合物处理后bcr/ablmrna表达量降低,且具有一定的剂量依赖性。测试例2按照发明专利201910706129.7中实施例3的方法,测试式a所示结构的雄甾烷衍生物和式b所示结构的雄甾烷衍生物对hedgehog信号通路的抑制作用,并与发明专利201910706129.7中式i所示结构的化合物进行比较,具体如下:小鼠报告细胞系-10t1/2-gliluc[s12]细胞(源自细胞系c3h10t1/2atcc#ccl-226);小鼠胚胎成纤维细胞;生长培养基:用10%胎牛血清(fbs)、10单位/ml青霉素、100μg/ml链霉素、2mm谷氨酰胺和10mmhepes的dulbecco改进的eagles培养基(dmem)。人报告细胞系-hepm-gliluc[mz24]-细胞(源自hepm,人胚胎腭突间充质atcc#crl-1486);生长培养基:用10~20%胎牛血清(fbs)、10单位/ml青霉素、100μg/ml链霉素、2mm谷氨酰胺和10mmhepesph7.2的极限必需培养基(mem;包含earle's盐)。微量滴定板(mtp):将细胞铺在96孔mtp(白色,平底,视野透明)中用于荧光素酶测定。荧光素酶检测用培养基:用0.5%fbs、10单位/ml青霉素、100μg/ml链霉素、2mm谷氨酰胺和10mmhepesph7.2的dmem。pbs/ca/mg混合物:含有0.5mmcacl2和1mmmgcl2的磷酸缓冲盐溶液(pbs)。测定程序:将s12和mz24细胞分别接种于各自的培养基中,并置于37℃、5%co2的培养箱进行培养。所述s12和mz24细胞分别含有被hedgehog反应性gli启动子驱动的荧光素酶报告基因。每3天使细胞培养物以亚融合状态(sub-confluency)进行传代,其中,s12与细胞培养物的质量比为1:(20~40);mz24与细胞培养物的质量比为1:(3~10)。收获细胞,用生长培养基稀释,以10,000~20,000个细胞(s12)/100μl/孔或20,000~30,000个细胞(mz24)/100μl/孔接种于微量滴定板中,并在37℃和5%co2的培养箱中将细胞进一步温育约48h。在48h的温育后,针对s12细胞系,用包含s12和具有式i所示结构的化合物的荧光测定培养基、包含s12和vismodegib(一种经fda批准的hedgehog通路抑制剂)的荧光测定培养基、以及只包含s12的荧光测定培养基(100μl/孔)分别代替微量滴定板中的生长培养基,各培养基中s12细胞系变化浓度为0.1~0.3μg/ml。针对mz24细胞系,用包含mz24和具有式a(或式b)所示结构的化合物的荧光测定培养基、包含mz24和vismodegib(一种经fda批准的hedgehog通路抑制剂)的荧光测定培养基以及只包含mz24的荧光测定培养基(100μl/孔)分别代替微量滴定板中的生长培养基,各培养基中mz24细胞系变化浓度为0.5~1.0μg/ml。将细胞进一步温育24h。然后用荧光素酶报告基因检测试剂盒(luclitetm)对微量滴定板进行检测,测试不同浓度下一系列化合物对特定靶点的活性,然后拟合成曲线,按照本领域公知的ic50的计算方法得出ic50值。测定过程对厂商的方法进行了改良,其中去除培养基并用1:1的pbs/ca/mg:裂解缓冲液重溶底物,而不是直接用裂解缓冲液重溶底物。简言之,按体积比1:1将pbs/ca/mg与裂解缓冲液混合,向(1000份测定试剂盒)每个底物瓶中添加10ml。然后弃去来自微量滴定板的测定培养基,向每个孔中添加100μl该底物混合物。将板在室温下温育30min,然后用topcount读出器(packard)或analyst读出器(moleculardevices)测定代表荧光素酶报告基因相对表达水平的相对光单位(rlus)。并计算对应化合物的ic50值。表4为根据上述程序使用小鼠[s12]和人[mz24]gli报告细胞系测得的具体化合物的抑制hedgehog途径信号传导的ic50值。表4试验结果化合物ic50(s12)ic50(mz24)ic50均值(μm)具有式a所示结构的化合物0.0160.0210.019具有式b所示结构的化合物0.0160.0200.018vismodegib0.0200.0240.022根据表4中的数据可知,本发明提供的具有式a和式b所示结构的化合物同样具有hedgehog途径信号抑制作用,并且其ic50达到了与vismodegib相当的水平。由以上测试例可知,本发明提供的具有式a或式b所示结构的雄甾烷衍生物对于bcr-abl酪氨酸激酶及对应mrna水平具有非常好的抑制作用,并且所述雄甾烷衍生物对于hedgehog信号通路同样具有非常好的抑制作用,说明其对慢性髓性白血病具有明显的治疗效果。以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1