5-卤代-4硫-2的制作方法
5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物合成方法
技术领域
[0001]
本发明属于化学合成技术领域,本发明涉及5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的高效合成方法。
背景技术:
[0002]
1988年,elion和hitchings因“发现了药物治疗的重要原则”获得诺贝尔生理学或医学奖,这些原则促进了一系列新的核酸衍生物的开发(the nobel prize in physiology or medicine 1988-press release.nobel media ab[eb/ol].http://www.nobelprize.org/nobel_prizes/medicine/laureates/1988/press.html.),其中他们开发的6-硫鸟嘌呤(6-thioguanine,6-tgua)、6-巯基嘌呤(6-mercaptopurine,6-mp)和硫唑嘌呤(azathioprine)核酸衍生物在治疗白血病和抑制移植器官排斥等疾病中表现出出色的化疗效果(elion g b.the purine path to chemotherapy[j].science,1989,244(4900):41-47.)。硫化后的天然核苷化合物与天然核苷相比,在生物活性方面表现出独特的性质,研究表明含有硫羰基的核苷类化合物其嘧啶碱基上4-位羰基氧被硫取代后吸光度变大,最大吸收峰红移,对光极其敏感(霍书华,李德鹏,王健,等.分子科学学报,2013,29(5):392-396.),使dna分子对长波紫外线(uva)或近紫外线光敏感。其中4-硫代脱氧核苷类似物对紫外线很敏感,可作为潜在抗肿瘤药物,尤其是与近紫外光(uva)结合后可用于治疗皮肤癌(pridgeon sw,heer r,taylor gabr.j.cancer 2011,104,1869.)。
[0003]
基于硫代核苷这些独特的生物学性质,激发了科学家对新型硫代核苷化合物合成的兴趣。
[0004]
对于新化合物5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的合成目前还未有报道。在合成新化合物5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的过程中,尝试使用过去常用的硫化手段,例如使用劳森试剂硫化合成、硫代乙酸硫化合成、五硫化二磷硫化合成。虽常用的三种硫化手段都有新化合物5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的生成,但反应过程存在一定的缺陷。其中,1)劳森试剂硫化法存在反应温度过高(90℃左右),后处理困难,难以通过柱层析得到纯产物;2)硫代乙酸硫化法存在反应时间过长(通常过夜),硫代乙酸味道极大,后处理复杂,尤其柱层析时,会分离得到难闻的副产物。此外当硫代乙酸做硫化剂合成新化合物5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物时,发现产率较低;3)五硫化二磷硫化法存在反应温度过高等缺点。
技术实现要素:
[0005]
为了克服现有技术的不足,本发明提供一种5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的高效合成方法。该合成方法条件温和,反应高效,后处理简单。
[0006]
本发明的上述目的是通过以下技术方案实现的:
[0007]
5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物;具有通式(c)结构:
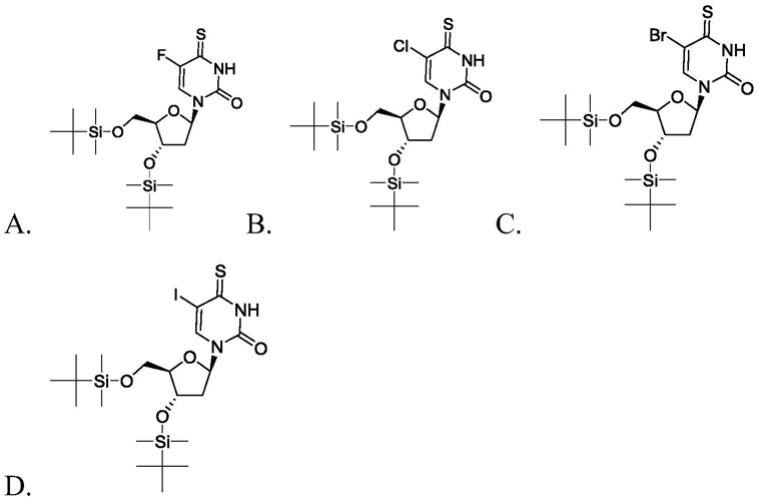
[0008][0009]
其中,x=f、cl、br、i。
[0010]
上述5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的具体结构式如下:
[0011][0012]
其中a为5-氟-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基脱氧尿苷,b为5-氯-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基脱氧尿苷,c为5-溴-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基脱氧尿苷,d为5-碘-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基脱氧尿苷。
[0013]
5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的高效合成方法;以5-卤代脱氧尿苷为原料,以nahs为硫化剂,在丙酮:水(v:v=3:1)的溶液中发生化学反应,最终得到通式(c)所示的化合物c;
[0014]
[0015]
其中,x=f、cl、br、i。
[0016]
进一步的,上述5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的高效合成方法步骤为:在氮气保护下,将5-卤代脱氧核苷化合物与叔丁基二甲基氯硅烷(tbscl)反应,反应完成后进行后处理得通式(a)所示的化合物a;化合物a接着与1,2,4-三唑溶解在无水乙腈溶剂中反应,得通式(b)所示的浅黄色固体化合物b;最后化合物b与nahs在丙酮:水(v:v=3:1)溶液中反应,得到通式(c)所示的黄色固体化合物c。
[0017]
进一步的,上述5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的高效合成方法具体步骤为:
[0018]
s1.化合物a的合成:在氮气保护下,将1当量5-取代脱氧核苷化合物和3当量咪唑溶解在二氯甲烷中,冰水浴下(0℃)搅拌30分钟;30分钟后,向反应体系中加入2.5当量叔丁基二甲基氯硅烷(tbscl),室温搅拌1小时;tlc检测反应结束后,用水进行猝灭,二氯甲烷萃取;合并有机相,用饱和nacl洗涤,无水naso
4
干燥,过滤,减压蒸去溶剂,得化合物a,为白色固体,无须纯化直接进行下一步s2;
[0019]
s2.化合物b的合成:将14当量的1,2,4-三唑溶解在无水乙腈中,冰水浴(0℃)搅拌下缓慢加入三氯氧磷,三乙胺;反应1小时后,投入上述s1制备的1当量白色固体化合物a,室温下搅拌过夜;tlc检测反应结束后,将反应液过滤,有机相用二氯甲烷稀释,饱和nahco
3
溶液洗涤,饱和nacl溶液洗涤,无水na
2
so
4
干燥,过滤,减压蒸去溶剂,得化合物b,为浅黄色固体,无须纯化直接进行下一步s3;
[0020]
s3.化合物c的合成:将1当量的化合物b溶解在丙酮:水(v:v=3:1)的溶液中,然后加入2当量的nahs,室温下搅拌30分钟左右,tlc检测反应结束,反应结束后加入水稀释,二氯甲烷萃取,饱和nacl溶液洗涤直至将反应溶液洗至中性,无水na
2
so
4
干燥,过滤,减压蒸去溶剂,及得到较纯黄色固体化合物c,更纯化合物c通过柱层析快速纯化得到。
[0021]
本发明与现有技术相比的有益效果是:
[0022]
本发明为制备此类5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物提供了一种高效的合成方法,此类5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物是首次合成。在从化合物a-化合物c的合成中,利用nahs作为硫化剂,丙酮:水(v:v=3:1)作为反应溶剂,在丙酮和水组成的混合溶剂中nahs解离成hs-离子,通过hs-作为亲核试剂进攻化合物b嘧啶环4位碳上的三唑基团,从而实现了高效快速的合成了一类新化合物5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物。该合成方法在后处理过程中利用nahs和反应溶剂易溶于水,而反应终产物难溶于水易溶于二氯甲烷的溶解性特点,简化了后处理过柱的纯化步骤。此外反应过程中避免使用现有技术常用的硫化试剂—劳森试剂和硫代乙酸。其中劳森试剂作为硫化试剂使用时,反应条件苛刻,温度常在90℃左右,后处理复杂,产物分离过程中伴有大量黑色副产物生成,反应完成时间一般在3-4小时。而硫代乙酸做硫化试剂使用时,虽然反应温度是室温,但硫代乙酸味道极大,产率低,后处理困难,且反应时间通常过夜。而本发明提供了一种室温下反应(26℃-30℃)进行,反应过程味道极小,反应迅速(通常30分钟内完成),产率高,后处理简单的一类新化合物5-卤代-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基核苷化合物的高效合成方法。
附图说明
[0023]
图1为本发明实施例1的化合物a
1
的核磁表征图谱。
[0024]
图2为本发明实施例1的化合物b
1
的核磁表征图谱。
[0025]
图3为本发明实施例1的化合物c
1
的核磁表征图谱。
具体实施方式
[0026]
下面通过具体实施例详述本发明,但不限制本发明的保护范围。如无特殊说明,本发明所采用的实验方法均为常规方法,所用实验器材、材料、试剂等均可从商业途径获得。
[0027]
实施例1
[0028]
5-氟-4硫-2
’
,3
’-
o-二叔丁基二甲硅烷基脱氧尿苷,其结构式为通式(c
1
)所示,
[0029][0030]
s1.化合物a
1
的合成:在氮气保护下,将脱氧胸苷(5g)和咪唑(4.15g)溶解在二氯甲烷(34ml)中,冰水浴下搅拌30分钟。30分钟后,向反应体系中加入叔丁基二甲基氯硅烷(tbscl)(7.65g),室温搅拌1小时。tlc检测反应结束后,用水进行猝灭,二氯甲烷萃取。合并有机相,用饱和nacl洗涤,无水naso
4
干燥,过滤,减压蒸去溶剂,得化合物a
1
,为白色固体,9.54g,产率99%,无须纯化直接进行下一步s2。
1
h nmr(cdcl
3
,500mhz)δ:8.62(s,1h),8.07(d,j=6.2hz,1h),6.30(t,j=6.0hz,1h),4.41(dt,j=6.2,3.1hz,1h),3.94(d,j=12.5hz,2h),3.77(d,j=11.1hz,1h),2.32(ddd,j=13.2,6.1,3.5hz,1h),2.06(p,j=6.3hz,1h),0.91(d,j=19.1hz,18h),0.11(dd,j=24.6,2.9hz,12h)。
[0031]
s2.化合物b
1
的合成:将1,2,4-三唑(6.44g)溶解在无水乙腈(41.05ml)中,冰水浴搅拌下缓慢加入三氯氧磷(153.33ml),三乙胺(101.19ml)。反应1小时后,投入上述s1制备的化合物a
1
白色固体(3g),室温下搅拌过夜。tlc检测反应结束后,将反应液过滤,有机相用二氯甲烷稀释,饱和nahco
3
溶液洗涤,饱和nacl溶液洗涤,无水na
2
so
4
干燥,过滤,减压蒸去溶剂,得化合物b
1
,为浅黄色固体,3.28g,产率99%,无须纯化直接进行下一步s3。
1
h nmr(cdcl
3
,500mhz)δ:9.26(s,1h),8.86(d,j=6.0hz,1h),8.22(s,1h),6.23(d,j=5.3hz,1h),4.41(d,j=4.8hz,1h),4.15
–
3.97(m,3h),3.83(d,j=11.5hz,1h),2.61(dq,j=13.2,6.3,5.2hz,1h),2.23(dt,j=13.5,5.0hz,1h),2.05(s,0h),0.94(s,9h),0.89(s,9h),0.15(d,j=9.9hz,6h),0.08(s,6h)。
[0032]
s3.化合物c
1
的合成:室温下,将化合物b1(300mg)溶解在1.15ml的丙酮:水(v:v=3:1)组成的溶液中,待充分溶解后加入64.5mg的nahs,反应溶液由开始的浑浊液变为透明澄清浅黄色溶液最后变为深棕色透明溶液,大约30分钟后通过tlc(pe:ea=3:1)检测,发现反应完成。向反应溶剂中加入水稀释,有大量黄色固体生成(此黄色固体为杂质),然后用二
氯甲烷萃取,饱和nacl充分洗涤,无水na
2
so
4
干燥,减压蒸去溶剂,得到黄色油状固体c
1
,过夜固化成黄色糖浆,251.9mg,产率90%。
1
h nmr(cdcl
3
,500mhz)δ:9.50(s,1h),8.05(d,j=3.9hz,1h),6.23(d,j=5.4hz,1h),4.42(s,1h),3.99
–
3.66(m,3h),2.39
–
2.24(m,1h),2.09(dt,j=12.9,5.7hz,1h),0.91(d,j=18.8hz,18h),0.17
–-
0.00(m,12h)。
[0033]
以上所述实施方式仅为本发明的优选实施例,而并非本发明可行实施的全部实施例。对于本领域一般技术人员而言,在不背离本发明原理和精神的前提下对其所作出的任何显而易见的改动,都应当被认为包含在本发明的权利要求保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1