一种磺达肝癸钠杂质的制备方法与流程
1.本发明涉及一种磺达肝癸钠杂质的制备方法,属于医药技术领域。
背景技术:
2.磺达肝癸钠为赛诺菲和欧加农公司共同开发的新一代抗血栓制剂,为人工合成的戊糖类物质,它是特异性地抑制血液凝集级联中的xa因子而最终显现抗凝血效应。磺达肝癸钠化学名称为:甲基o-(2-脱氧-6-o-磺酸基-2-磺酰胺基-α-d-吡喃葡萄糖)-(1
→
4)-o-(β-d-吡喃葡萄糖醛酸)-(1
→
4)-o-(2-脱氧-3,6-o-二磺酸基-2-磺酰胺基-α-d-吡喃葡萄糖)-(1
→
4)-o-(2-o-磺酸基-α-l-吡喃艾杜糖醛酸)-(1
→
4)-2-脱氧-6-o-磺酸基-2-磺酰胺基-α-d-吡喃葡萄糖苷十钠盐,其结构式如下所示:
[0003][0004]
磺达肝癸钠和at iii特异性结合,并抑制凝血因子xa的活性,防止血栓的形成。血液清除t1/2为13~21h,每日注射1次即可。一般患者注射时不需要进行常规凝血监测。磺达肝癸钠注射剂于2001年12月获得ema批准,2002年初获美国fda批准上市。
[0005]
磺达肝癸钠杂质化合物ⅰ化学名称为甲基(2-脱氧-2-氨基磺酸钠-6-o-磺酸钠-α-d-吡喃葡萄糖)-(1
→
4)-(β-d-吡喃葡萄糖醛酸钠)-(1
→
4)-(2-脱氧-2-氨基磺酸钠-3,6-二-o-磺酸钠-α-d-吡喃葡萄糖)-(1
→
4)-(2,3-二-o-磺酸钠-α-l-吡喃艾杜糖酸钠)-2-脱氧-2-氨基磺酸钠-6-o-磺酸钠-α-d-吡喃葡萄糖苷,其结构式为:
[0006][0007]
杂质化合物ⅰ在美国药典中,其相对于磺达肝癸钠的rrt为0.93。该杂质通常是从磺达肝癸钠反应液或粗品中分离纯化得到,但是该杂质在反应液或粗品中的含量较低,大量获得的难度较高,成本较大。而目前有关杂质化合物i的合成方法暂无报道,杂质的存在直接关系到药品的质量和安全,因此对其进行合成鉴定对产品的质量控制有重要的意义。
技术实现要素:
[0008]
本发明目的在于提供一种磺达肝癸钠杂质化合物ⅰ的制备方法,该工艺设计合理,反应过程稳定可控,得到的化合物纯度较高,对磺达肝癸钠及制剂的质量研究具有重要意义。
[0009]
本发明提供的磺达肝癸钠杂质化合物ⅰ的制备方法,包括以下步骤:
[0010]
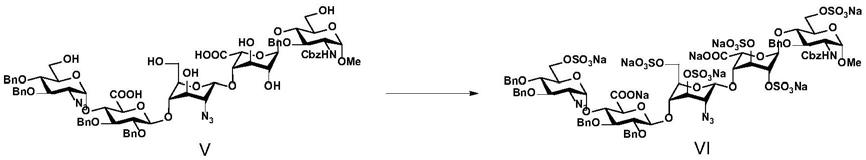
步骤1:化合物ⅳ的制备
[0011][0012]
化合物ⅱ和化合物ⅲ在偶联催化剂的作用下,反应得到化合物ⅳ;
[0013]
步骤2:化合物
ⅴ
的制备
[0014][0015]
化合物ⅳ在碱性条件下发生水解反应,再经盐酸调ph至酸性后得到化合物
ⅴ
;
[0016]
步骤3:化合物ⅵ的制备
[0017][0018]
化合物
ⅴ
与磺化试剂a发生反应生成硫酸酯类化合物,硫酸酯类化合物经钠离子交换后得到化合物ⅵ;
[0019]
步骤4:化合物ⅶ的制备
[0020][0021]
化合物ⅵ在催化剂作用下,发生还原和脱苄基反应,得到化合物ⅶ;
[0022]
步骤5:杂质化合物ⅰ的制备
[0023][0024]
化合物ⅶ与磺化试剂b发生反应生成磺酸酯类化合物,磺酸酯类化合物经钠离子交换后并纯化后得到杂质化合物ⅰ;
[0025]
化合物ⅱ或ⅳ中,所述的r1、r2、r3各自独立为苯甲酰基、烷基酰基、芳基酰基、烷基芳基酰基或被卤素取代的苯甲酰基、烷基酰基、芳基酰基或烷基芳基酰基,乙氧基羰基、叔丁氧基羰基、烯丙基氧基羰基、碳酸酯保护基中的任意一种。
[0026]
进一步,作为优选,步骤1中,所述的化合物ⅱ和化合物ⅲ及偶联催化剂的摩尔比为1:0.9~1.1:0.8~1.2。
[0027]
进一步,作为优选,步骤1中,所述的偶联催化剂为tbdmsotf、tmsotf、agotf中的一种或两种及以上的混合物。
[0028]
进一步,步骤1中,反应温度优选为-20~-40℃,反应时间优选为2~6h。
[0029]
进一步,作为优选,步骤2中,所述的碱性条件通过碱性物质提供,所述的碱性物质为氢氧化钠、氢氧化钾、氢氧化锂、碳酸钠中的一种或任意几种的混合;所述的反应温度10~60℃,优选20~30℃;水解反应采用的反应溶剂可以为甲醇、乙醇、四氢呋喃、二氧六环中的一种或几种与水的混合体系;所述反应还包括重结晶步骤,所述的重结晶体系可以为乙酸乙酯/正己烷、乙酸乙酯/石油醚。
[0030]
进一步,作为优选,步骤3中,所述的磺化试剂a可以为三甲基铵三氧化硫共聚物(so3nme3)、三氧化硫吡啶络合物(so3py)、氯磺酸、氨基磺酸中的一种或任意几种的混合;所述的磺化试剂a为化合物
ⅴ
的15~20摩尔当量。
[0031]
进一步,作为优选,步骤3中,所述的反应温度40~80℃,优选50~70℃。
[0032]
进一步,作为优选,步骤3中,所述的钠离子交换可以使用732阳离子交换树脂、
dowex 50wx4树脂,或氢氧化钠、碳酸钠中的一种或几种的混合使用。
[0033]
进一步,作为优选,步骤4中,所述的催化剂可以为钯炭、氢氧化钯、醋酸钯、钯黑中的一种或几种的混合;所述的化合物ⅵ与催化剂的摩尔比为1:0.1~2;反应溶剂可以为乙醇、甲醇、纯化水中的一种或几种的混合;反应温度可以为10~60℃,氢气压力为0.5~4mpa;为了提高制备过程中的安全性,优选纯化水或纯化水/醇大于70/30(v/v)作为反应溶剂,氢气压力为1.0~1.5mp,反应温度为20~30℃。
[0034]
进一步,作为优选,步骤5中,所述的磺化试剂b可以为三氧化硫及三氧化硫衍生物如三甲基铵三氧化硫共聚物(so3nme3)、三氧化硫吡啶络合物(so3py)、氯磺酸、氨基磺酸中的一种或几种的混合;优选,所述的磺化试剂b为化合物ⅶ的6~10摩尔当量;反应ph为8~12,优选9~10;反应温度为6~14℃,优选8~12℃;所述的交换过程采用交换树脂:732阳离子交换树脂、dowex 50wx4树脂,或氢氧化钠、碳酸钠中的一种或几种的混合使用。
[0035]
与现有技术相比,本发明的有益效果在于:
[0036]
本发明提供的合成方法操作和后处理简单,反应条件温和,工艺稳定,收率高,重现性好,弥补了该杂质i无制备方法的空白,为磺达肝癸钠的质量研究和控制奠定了良好的基础。
附图说明
[0037]
图1:杂质化合物ⅰ的ms(esi)谱图;
[0038]
图2:杂质化合物ⅰ的1h nmr谱图。
具体实施方式
[0039]
以下将结合较优的实施例对本发明作进一步的详细说明,但不作为本发明的限制。
[0040]
实施例1-6均采用化合物ⅱ合成得到化合物
ⅴ
:
[0041][0042]
实施例7-8均采用以下步骤合成得到化合物ⅵ:
[0043][0044]
实施例9-10均采用以下步骤合成得到化合物ⅶ:
[0045][0046]
实施例11-12均采用以下步骤合成得到化合物ⅰ:
[0047][0048]
实施例1:化合物
ⅴ
的制备
[0049]
将2.00g化合物
ⅱ-
1和3.00g化合物ⅲ溶于50ml无水二氯甲烷中,于-20℃滴加0.5ml tbdmsotf,继续反应2小时。于-20℃加入三乙胺搅拌淬灭2小时,过滤浓缩,所得样品以乙酸乙酯/正己烷(v/v=1/3)为洗脱剂经200-300目硅胶层析后得到2.34g化合物
ⅳ-
1,收率50.4%。
[0050]
将2.20g化合物
ⅳ-
1加入到44ml thf/h2o(v/v=2/1)中搅拌溶解,加入22ml 1n naoh溶液并于20℃反应12小时,反应完毕,用1n hcl调ph至3,用二氯甲烷萃取,有机相用饱和食盐水洗至中性,无水硫酸钠干燥并旋干,得到1.92g粗品。粗品用7.7ml乙酸乙酯溶解,滴加3.8ml正己烷,搅拌析晶3小时,过滤并干40℃真空干燥后得到1.46g化合物
ⅴ
,收率81.5%。
[0051]
实施例2:化合物
ⅴ
的制备
[0052]
将2.00g化合物
ⅱ-
1和2.90g化合物ⅲ溶于50ml无水二氯甲烷中,于-30℃滴加0.5ml tmsotf,继续反应5小时。于-30℃加入三乙胺搅拌淬灭2小时,过滤浓缩,所得样品以乙酸乙酯/正己烷(v/v=1/3)为洗脱剂经200-300目硅胶层析后得到2.16g化合物
ⅳ-
1,收率46.5%。
[0053]
将2.10g化合物
ⅳ-
1加入到42ml thf/h2o(v/v=2/1)中搅拌溶解,加入21ml 1n koh溶液并于25℃反应12小时,反应完毕,用1n hcl调ph至3,用二氯甲烷萃取,有机相用饱和食盐水洗至中性,无水硫酸钠干燥并旋干,得到1.71g粗品。粗品用6.8ml乙酸乙酯溶解,滴加3.4ml正己烷,搅拌析晶3小时,过滤并干40℃真空干燥后得到1.33g化合物
ⅴ
,收率77.7%。
[0054]
实施例3:化合物
ⅴ
的制备
[0055]
将8.50g化合物
ⅱ-
2和12.00g化合物ⅲ溶于205ml无水二氯甲烷中,于-20℃滴加2ml tbdmsotf,继续反应2小时。于-20℃加入三乙胺搅拌淬灭2小时,过滤浓缩,所得样品以
乙酸乙酯/正己烷(v/v=1/3)为洗脱剂经200-300目硅胶层析后得到10.61g化合物
ⅳ-
2,收率56.1%。
[0056]
将10g化合物
ⅳ-
2加入到200ml thf/h2o(v/v=2/1)中搅拌溶解,加入100ml 1n naoh溶液并于25℃反应12小时,反应完毕,用1n hcl调ph至3,用二氯甲烷萃取,有机相用饱和食盐水洗至中性,无水硫酸钠干燥并旋干,得到9.12g粗品。粗品用36.4ml乙酸乙酯溶解,滴加18.2ml正己烷,搅拌析晶3小时,过滤并在40℃真空干燥后得到6.60g化合物
ⅴ
,收率83.7%。
[0057]
实施例4:化合物
ⅴ
的制备
[0058]
将4.00g化合物
ⅱ-
2和6.00g化合物ⅲ溶于100ml无水二氯甲烷中,于-20℃滴加1ml tbdmsotf,继续反应4小时。于-20℃加入三乙胺搅拌淬灭2小时,过滤浓缩,所得样品以乙酸乙酯/正己烷(v/v=1/3)为洗脱剂经200-300目硅胶层析后得到4.65g化合物
ⅳ-
2,收率52.2%。
[0059]
将4.6g化合物
ⅳ-
2加入到92ml thf/h2o(v/v=2/1)中搅拌溶解,加入46ml 1n naoh溶液并于25℃反应12小时,反应完毕,用1n hcl调ph至3,用二氯甲烷萃取,有机相用饱和食盐水洗至中性,无水硫酸钠干燥并旋干,得到4.3g粗品。粗品用17.2ml乙酸乙酯溶解,滴加8.6ml正己烷,搅拌析晶3小时,过滤并在40℃真空干燥后得到3.06g化合物
ⅴ
,收率84.4%。
[0060]
实施例5:化合物
ⅴ
的制备
[0061]
将1.00g化合物
ⅱ-
3和1.50g化合物ⅲ溶于25ml无水二氯甲烷中,于-20℃滴加0.25ml tbdmsotf,继续反应2小时。于-20℃加入三乙胺搅拌淬灭2小时,过滤浓缩,所得样品以乙酸乙酯/正己烷(v/v=1/3)为洗脱剂经200-300目硅胶层析后得到1.16g化合物
ⅳ-
3,收率50.0%。
[0062]
将1.10g化合物
ⅳ-
3加入到22ml thf/h2o(v/v=2/1)中搅拌溶解,加入11ml 1n naoh溶液并于20℃反应12小时,反应完毕,用1n hcl调ph至3,用二氯甲烷萃取,有机相用饱和食盐水洗至中性,无水硫酸钠干燥并旋干,得到0.90g粗品。粗品用3.6ml乙酸乙酯溶解,滴加1.8ml正己烷,搅拌析晶3小时,过滤并在40℃真空干燥后得到0.72g化合物
ⅴ
,收率80.4%。
[0063]
实施例6:化合物
ⅴ
的制备
[0064]
将1.00g化合物
ⅱ-
3和1.50g化合物ⅲ溶于25ml无水二氯甲烷中,于-20℃滴加0.25ml tbdmsotf,继续反应2小时。于-20℃加入三乙胺搅拌淬灭2小时,过滤浓缩,所得样品以乙酸乙酯/正己烷(v/v=1/3)为洗脱剂经200-300目硅胶层析后得到1.20g化合物
ⅳ-
3,收率51.7%。
[0065]
将1.10g化合物
ⅳ-
3加入到22ml thf/h2o(v/v=2/1)中搅拌溶解,加入11ml 1n naoh溶液并于20℃反应12小时,反应完毕,用1n hcl调ph至3,用二氯甲烷萃取,有机相用饱和食盐水洗至中性,无水硫酸钠干燥并旋干,得到0.94g粗品。粗品用3.6ml乙酸乙酯溶解,滴加1.8ml石油醚,搅拌析晶3小时,过滤并在40℃真空干燥后得到0.69g化合物
ⅴ
,收率77.0%。
[0066]
实施例7:化合物ⅵ的制备
[0067]
将6.50g化合物
ⅴ
,10g so3nme3加入到65ml dmf中,升温至60℃反应12小时,反应
完毕将反应液减压浓缩至油状物,油状物加入13ml二氯甲烷/甲醇溶解(v/v=1/1)溶解,所得溶液以二氯甲烷/甲醇(v/v=1/1)为洗脱剂经lh20层析除去无机盐,层析液减压浓缩后使用适量甲醇/水溶解(v/v=1/1)溶解,以甲醇/水(v/v=1/1)为洗脱剂经732阳离子交换树脂交换并浓缩得到8.02g化合物ⅵ,收率85.9%。
[0068]
实施例8:化合物ⅵ的制备
[0069]
将3.00g化合物
ⅴ
,5.00g so3nme3加入到32ml dmf中,升温至50℃反应12小时,反应完毕,加入ml 4n氢氧化钠溶液,搅拌2小时后过滤,将滤液减压浓缩至油状物,油状物加入10ml二氯甲烷/甲醇溶解(v/v=1/1)溶解,所得溶液以二氯甲烷/甲醇(v/v=1/1)为洗脱剂经lh20层析除去无机盐,层析液浓缩得到2.78g化合物ⅵ,收率64.5%。
[0070]
实施例9:化合物ⅶ的制备
[0071]
氢化釜中加入7.80g化合物ⅵ,5.6g 10%pd/c,156ml乙醇/纯化水(v/v=3/7),氢气压力1.0mpa,25℃反应48小时,反应完毕,过滤旋干得到3.81g化合物ⅶ,收率69.3%。
[0072]
实施例10:化合物ⅶ的制备
[0073]
氢化釜中加入2.70g化合物ⅵ,0.2g pd(oac)2,54ml纯化水,氢气压力1.5mpa,40℃反应64小时,反应完毕,过滤旋干得到1.30g化合物ⅶ,收率68.3%。
[0074]
实施例11:杂质化合物ⅰ的制备
[0075]
将3.70g化合物ⅶ溶于74ml纯化水中,控制温度9-10℃,用2n naoh调溶液ph至9-10,缓慢加入3.70g so3py,维持ph9-10反应6小时,反应完毕,将反应液以纯化水为洗脱剂经g25层析并减压浓缩至干得到杂质化合物ⅰ粗品。粗品经柱分离纯化得到1.78g杂质化合物ⅰ,收率40.1%,纯度97.1%。
[0076]
实施例12:杂质化合物ⅰ的制备
[0077]
将1.00g化合物ⅶ溶于20ml纯化水中,控制温度8-9℃,用2n naoh调溶液ph至10-11,缓慢加入0.8g so3py,维持ph10-11反应6小时,反应完毕,将反应液以纯化水为洗脱剂经g25层析并减压浓缩至干得到杂质化合物ⅰ粗品。粗品使用适量甲醇/水溶解(v/v=1/1)溶解,以甲醇/水(v/v=1/1)为洗脱剂经732阳离子交换树脂交换浓缩后再经柱分离纯化得到0.43g杂质化合物ⅰ,收率35.8%,纯度大于96.7%。
[0078]
化合物ⅰ的分子结构信息:
[0079]
分子式:c
31h42
n3na
11o52
s9[0080]
exact mass:1828.7095
[0081]
ms(esi)m/z:937.3467=[(m+2na)/2]
+
,
[0082]
926.3549=[(m+na+h)/2]
+
[0083]1h nmr(d2o,ppm)δ:5.52(d,1h),5.25(s,1h),5.23(d,1h),4.91(d,1h),4.83(s,1h),4,75(s,1h),4.54(d,1h),4.42-4.24(m,5h),4.20-4.10(m,3h),4.06(d,3h),3.95-3.80(m,3h),3.80-3.65(m,4h),3.65-3.55(m,2h),3.55-3.45(m,2h),3.33(dd,1h),3.35-3.25(m,4h),3.20-3.10(m,2h)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1