siRNA的制备方法
本申请属于核酸提取技术领域,尤其涉及一种sirna的制备方法。
背景技术:
rna干扰(rnainterference,rnai)是近年发展起来的一种阻抑基因表达的新方法,该技术可以通过双链rna的介导,降解靶基因的mrna,在转录后水平上特异性地阻断或降低靶基因的表达,达到类似基因敲除的效果,而操作比基因敲除简单,有着投入少、实验周期短等优势,是一种可以大量进行基因功能检测的高效且经济的研究方法,已经被广泛用于研究特定基因的功能。
在rnai实验中,获得小干扰rna(smallinterferingrna,sirna)是沉默靶基因的首要步骤。目前有多种方法制备sirna,其中较为常用的有化学合成法、体外转录法、体内载体表达法、sirna表达框架法、rnaseⅲ体外消化dsrna(double-strandedrna,双链rna)法等。
一个基因的不同sirna对其可能会有显著不同的抑制效果,化学合成法、体外转录法、体内载体表达法、sirna表达框架法等均需要寻找和验证最有效的sirna,因而可能会消耗更多时间。而使用rnaseⅲ消化长dsrna制备法无需验证和寻找有效地特定sirna,可以很好地解决这个问题,为研究者节省不少经费及时间。该方法主要步骤为:先通过pcr制备dna模板(在pcr引物地5’端加上t7启动子序列,以启动转录),使用加上t7启动子序列的dna模板体外转录制备dsrna,然后使用大肠杆菌rnaseⅲ消化dsrna,形成sirna片段,去除未消化的dsrna即可纯化得到sirna混合物。该方法得到的产物含有多种不同序列的sirna,不同sirna可能存在序列的重叠,几乎可以覆盖整个靶区段,不必筛选有效片段即可保证可有效降解靶mrna,适合快速经济地研究某一基因功能缺失的表型。然而rnaseⅲ消化长dsrna除了产生12-30bp的短的dsrna,也产生一些>30bp的dsrna。在哺乳动物细胞中,长度超过30bp的长dsrna不仅无法触发细胞中的rnai,而且会激活通常由ifn诱导的dsrna依赖的激酶pkr和2'-5'-寡腺苷酸合成酶。激活的pkr通过磷酸化翻译因子真核起始因子2α来抑制翻译过程,而2'-5'-寡腺苷酸合成酶则通过激活rnasel引起非特异性mrna降解,从而对细胞会产生毒性作用。因此,需要对rnaseⅲ消化后的产物进行纯化,以获得较纯的12-30bp的sirna。
dsrna的分子量=碱基数(bp)x680da/bp,那么30bpdsrna对应的分子量即为20.4kda。因此,目前普遍采用30kda的超滤管纯化rnaseⅲ消化后的dsrna混合物。然而,超滤管价格昂贵,不适用于需要研究多个基因的大量制备sirna;而且超滤管纯化在上样前需要将rnaseⅲ消化的混合物稀释,导致纯化的sirna浓度较低,需要再次进行sirna浓缩步骤,操作较繁琐。
因此,相关技术有待改进。
技术实现要素:
本申请的目的在于提供一种sirna的制备方法,旨在解决现有rnaseⅲ体外消化dsrna制备sirna成本高、步骤繁琐的技术问题。
为实现上述申请目的,本申请采用的技术方案如下:
本申请提供一种sirna的制备方法,其特征在于,包括如下步骤:
体外转录获得目的基因的dsrna;
用rnaseⅲ对所述目的基因的dsrna进行消化处理,得到消化产物;
将所述消化产物凝胶电泳后,切割核酸片段<30bp的胶块;
将所述胶块研磨冻融后,获得sirna。
本申请构建一种简单的sirna的制备方法,该制备方法是对现有rnaseⅲ体外消化dsrna法进行改进,具体地,该制备方法基于rnaseⅲ消化体外转录获得目的基因的dsrna,然后对消化产物凝胶电泳以切割核酸片段<30bp的胶块,通过对该胶块研磨冻融后,释放出sirna。该制备方法可以有效去除rnaseⅲ消化产物中>30bp的dsrna片段,具有准确有效、操作简便、成本低廉的特点,为纯化sirna以及rnai研究带来极大的方便,具有很好的应用前景。
附图说明
为了更清楚地说明本申请实施例中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本申请的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
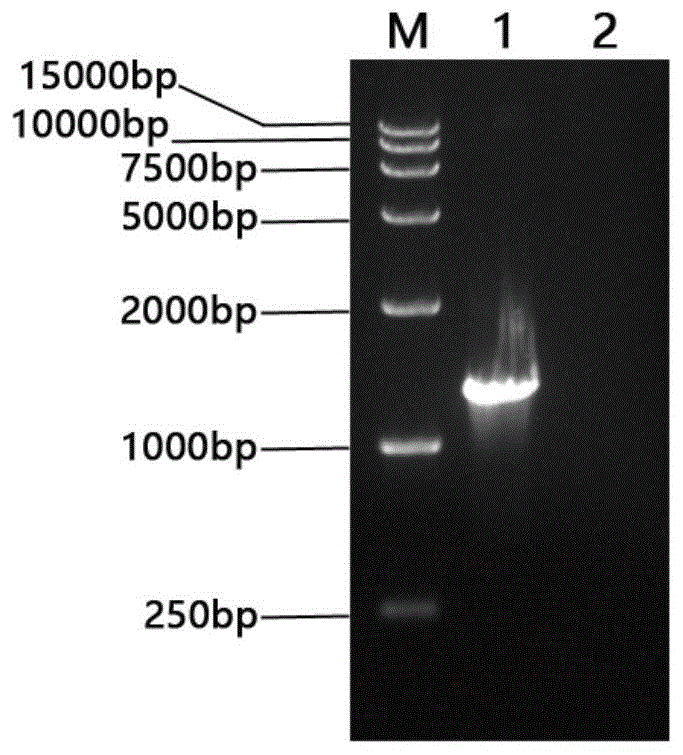
图1是本申请实施例提供的以含t7启动子序列的完整引物扩增unigene7122含t7启动子序列dna的琼脂糖凝胶电泳图;其中:m.dl15000dnamarker,1.含t7启动子序列的unigene7122扩增结果,2.阴性对照;
图2是本申请实施例提供的以含t7启动子序列的完整引物扩增unigene12835、unigene19308含t7启动子序列dna的琼脂糖凝胶电泳图;其中,m.dl2000dnamarker;1.阴性对照(t7-u12835f1、t7-u12835r1),2.含t7启动子序列的unigene12835(t7-u12835f1、t7-u12835r1)扩增结果,3.阴性对照(t7-u12835f2、t7-u12835r2),4.含t7启动子序列的unigene12835(t7-u12835f2、t7-u12835r2)扩增结果,5.阴性对照(t7-u19308f1、t7-u9308r1),6.含t7启动子序列的unigene19308(t7-u19308f1、t7-u9308r1)扩增结果,7.阴性对照(t7-u19308f2、t7-u9308r2),8.含t7启动子序列的unigene19308(t7-u19308f2、t7-u9308r2)扩增结果;两个基因后续实验分别使用引物t7-u12835f1、t7-u12835r1和t7-u19308f1、t7-u9308r1;
图3是本申请实施例提供的以含t7启动子序列的完整引物扩增unigene13423等5个基因含t7启动子序列dna的琼脂糖凝胶电泳图;其中:m.dl2000dnamarker,1.含t7启动子序列的unigene13423扩增结果,2.含t7启动子序列的cl1989.contig1扩增结果,3.含t7启动子序列的cl3290.contig2扩增结果,4.含t7启动子序列的cl3871.contig2扩增结果,5.含t7启动子序列的cl51.contig2扩增结果;
图4是本申请实施例提供的unigene12835等8个基因的dsrna及酶切后sirna产物的琼脂糖凝胶电泳图;其中:m1.dl2000dnamarker,m2.20bpdnaladdermarker,1.unigene12835dsrna,2.unigene12835sirnamix,3.unigene13423dsrna,4.unigene13423sirnamix,5.unigene19308dsrna,6.unigene19308sirnamix,7.unigene7122dsrna,8.unigene7122sirnamix,9.cl1989.contig1dsrna,10.cl1989.contig1sirnamix,11.cl3290.contig2dsrna,12.cl3290.contig2sirnamix,13.cl3871.contig1dsrna,14.cl3871.contig1sirnamix,15.cl51.contig3dsrna,16.cl51.contig3sirnamix;
图5是本申请实施例提供的unigene12835、unigene19308基因的sirna纯化产物的琼脂糖凝胶电泳图;其中,m.dl5000dnamarker,1.unigene12835未纯化的sirnamix,2.unigene12835纯化含goldview的sirna,3.unigene12835纯化不含goldview的sirna,4.unigene19308未纯化的sirnamix,5.unigene19308纯化含goldview的sirna,6.unigene19308纯化不含goldview的sirna;
图6是本申请实施例提供的cl3290.contig2、cl51.contig2基因的sirna纯化产物的琼脂糖凝胶电泳图;其中,m.20bpdnaladdermarker,1.cl3290.contig2未纯化的sirnamix,2.cl3290.contig2纯化含goldview的sirna,3.cl3290.contig2纯化不含goldview的sirna,4.cl51.contig3未纯化的sirnamix,5.cl51.contig3纯化含goldview的sirna,6.cl51.contig3纯化不含goldview的sirna;
图7是本申请实施例提供的unigene13423基因的sirna纯化产物的琼脂糖凝胶电泳图;其中,m.20bpdnaladdermarker,1.unigene13423未纯化的sirnamix,2.unigene13423纯化含goldview的sirna,3.unigene13423纯化不含goldview的sirna;
图8是本申请实施例提供的unigene12835经过靶向干扰后的基因相对表达量(以β-actin为内参基因);其中,(a)以nc组为对照的相对表达量,(b)以mock组为对照的相对表达量;
图9是本申请实施例提供的unigene19308经过靶向干扰后的基因相对表达量(以β-actin为内参基因);其中,(a)以nc组为对照的相对表达量,(b)以mock组为对照的相对表达量;
图10是本申请实施例提供的cl51.contig3经过靶向干扰后的基因相对表达量(以β-actin为内参基因);其中,(a)以nc组为对照的相对表达量,(b)以mock组为对照的相对表达量;
图11是本申请实施例提供的unigene13423经过靶向干扰后的基因相对表达量(以β-actin为内参基因);其中,(a)以nc组为对照的相对表达量,(b)以mock组为对照的相对表达量;
图12是本申请实施例提供的cl3290.contig2经过靶向干扰后的基因相对表达量(以β-actin为内参基因);其中,(a)以nc组为对照的相对表达量,(b)以mock组为对照的相对表达量;
图13是用rnaseⅲ对cl3290.contig2dsrna消化后未纯化的cl3290.contig2sirnamix与本申请实施例提供的经纯化后的cl3290.contig2sirna对目的基因cl3290.contig2经过靶向干扰后的基因相对表达量(以β-actin为内参基因)对比图;其中,(a)以nc组为对照的相对表达量,(b)以mock组为对照的相对表达量;
图14是对比例1用30k超滤管纯化sirna的琼脂糖凝胶电泳图;其中,m.20bpdnaladdermarker,1.unigene13423dsrna,2.unigene13423未纯化的sirnamix,3.unigene13423sirnamix经30k超滤管流出样,4.unigene13423sirnamix经30k超滤管截留样,5.unigene7122dsrna,6.unigene7122未纯化的sirnamix,7.unigene7122sirnamix经30k超滤管流出样,8.unigene7122sirnamix经30k超滤管截留样。
具体实施方式
为了使本申请要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本申请进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本申请,并不用于限定本申请。
本申请实施例提供一种sirna的制备方法,该制备方法包括如下步骤:
s01:体外转录获得目的基因的dsrna;
s02:用rnaseⅲ对所述目的基因的dsrna进行消化处理,得到消化产物;
s03:将所述消化产物凝胶电泳后,切割核酸片段<30bp的胶块;
s04:将所述胶块研磨冻融后,获得sirna。
本申请构建一种简单的sirna的制备方法,该制备方法是对现有rnaseⅲ体外消化dsrna法进行改进,具体地,该制备方法基于rnaseⅲ消化体外转录获得目的基因的dsrna,然后对消化产物凝胶电泳以切割核酸片段<30bp的胶块,通过对该胶块研磨冻融后,释放出sirna。该制备方法可以有效去除rnaseⅲ消化产物中>30bp的dsrna片段,具有准确有效、操作简便、成本低廉的特点,为纯化sirna以及rnai研究带来极大的方便,具有很好的应用前景。
步骤s01中,所述体外转录获得目的基因的dsrna的步骤包括:将总rna反转录得到cdna,用含有t7启动子的引物扩增所述cdna,然后进行体外转录,并用核酸酶消化。通过不同的含有t7启动子的引物设计,可以扩增所述cdna中不同目的基因,从而最终可以制备得到针对该目的基因的sirna。总rna的提取可以使用takara公司的rnaisoplus试剂盒进行;反转录可以通过takara公司的试剂盒primescripttm1ststrandcdnasynthesiskit进行;含有t7启动子的引物可以自行设计,可以先使用软件primerpremier5设计各基因的引物,然后在正反向引物的5’端分别加上t7启动子序列,作为完整的引物扩增所研究的目的基因部分长度片段。本实施例中添加的t7启动子序列为:5'-taatacgactcactatagggaga-3'。在本申请实施例中,目的基因选用了unigene12835,unigene13423,unigene19308,unigene7122,cl1989.contig1,cl3290.contig2,cl3871.contig1,cl51.contig3等。
本申请中,为验证该sirna的提取效果,对鲤鱼上皮瘤细胞(epc)进行转录组测序,从中获得基因转录本(mrna)的部分序列编号以及功能注释:具体地,编号unigene12835预测为4f2cell-surfaceantigenheavychain-like[sinocyclocheilusrhinocerous],编号unigene19308预测为lysosomemembraneprotein2-like[sinocyclocheilusrhinocerous],编号unigene13423预测为niemann-pickc1protein-like[sinocyclocheilusrhinocerous],编号unigene7122预测为aminopeptidasen-like[sinocyclocheilusrhinocerous],编号cl1989.contig1为neuralcelladhesionmolecule1aprecursor[daniorerio],编号cl3290.contig2为erythrocyteband7integralmembraneproteinisoformx2[daniorerio],编号cl3871.contig1为sourceofimmunodominantmhc-associatedpeptides[ctenopharyngodonidella],编号cl51.contig3预测为predicted:transferrinreceptorprotein1-like[sinocyclocheilusanshuiensis],以上述为目的基因进行对应的sirna的提取。
在步骤s02中的所述消化处理中,rnaseⅲ浓度为0.5-1.5u/μl,优选0.5-1.5u/μl,这样浓度的消化反应体系消化效果更好。并通过rnaseⅲ消化dsrna获得片段较小的,包括12-30bpsirna的dsrna混合物。
在步骤s03中,所述凝胶电泳是浓度为3%的琼脂糖凝胶电泳。紫外下切下核酸片段<30bp的sirna的胶块,从而可以有效去除rnaseⅲ消化产物中>30bp的dsrna片段,然后经过研磨冻融即可释放出针对目的基因的sirna。
进一步地,所述研磨冻融的步骤包括:将所述胶块切碎后,依次冰冻和解冻融化,然后加水研磨。其中,所述冰冻的温度为-80℃~0℃,所述解冻融化的温度为20℃-27℃,上述温度范围可以更好地进行冰冻和解冻融化。而研磨可以使用电动研磨枪彻底研磨。所述研磨冻融的步骤重复2-4次,这样可以彻底释放目的基因的sirna,以提高提取的浓度。
进一步地,所述加水研磨后,还包括离心取上清。反复冻融融化胶块,释放出sirna,离心取上清获得纯化的sirna。在一实施例中,所述目的基因为unigene12835,得到的所述sirna为unigene12835sirna;或者,所述目的基因为unigene13423,得到的所述sirna为unigene13423sirna;所述目的基因为unigene19308,得到的所述sirna为unigene19308sirna;所述目的基因为unigene7122,得到的所述sirna为unigene7122sirna;所述目的基因为cl1989.contig1,得到的所述sirna为cl1989.contig1sirna;所述目的基因为cl3290.contig2,得到的所述sirna为cl3290.contig2sirna;所述目的基因为cl3871.contig1,得到的所述sirna为cl3871.contig1sirna;所述目的基因为cl51.contig3,得到的所述sirna为cl51.contig3sirna。
通过上述制备方法,可以获得高纯度的sirna,后续可以将得到的sirna用于转染鲤鱼上皮瘤细胞以检测靶基因沉默效果;在一实施例中,上述制备方法所述获得sirna之后,还包括配制成浓度为0.1nmol/l-0.5nmol/l的所述sirna用于转染。本申请实施例将制备的sirna转染至鲤鱼上皮瘤细胞(epc),具体地,提取细胞rna,反转录成cdna,以cdna作为模板进行qrt-pcr反应,以β-actin作为内参,采用2-△△ct法计算目的基因相对表达量,结果发现sirna终浓度为0.5nmol/l时靶基因相对表达量下调50%左右。通过将终浓度为0.5nmol/l未纯化的cl3290.contig2sirnamix与纯化的cl3290.contig2sirna分别转染epc细胞,比较未纯化的cl3290.contig2sirnamix与纯化的cl3290.contig2sirna对靶基因沉默效果的差异,发现通过搅碎冻融法纯化的cl3290.contig2sirna对靶基因沉默效果明显高于未纯化的cl3290.contig2sirnamix,验证了搅碎冻融法纯化sirna的有效性。
本申请通过体外转录获得靶基因的dsrna,经rnaⅲ消化获得小片段的dsrna,通过切胶回收、搅碎冻融法纯化了rnaseⅲ消化的sirna混合物,得到较纯的sirna,并用纯化的sirna转染epc细胞检测靶基因沉默效果,发现sirna终浓度为0.5nmol/l时对靶基因表达下调50%左右。因此,本申请可以实现简便、低成本并且高效纯化sirna的目的,在rnai研究中具有重要的应用价值。
下面结合具体实施例进行说明。
实验材料与试剂
主要材料:鲤鱼上皮瘤细胞(epc);
主要试剂:sirnanc由广州锐博生物公司提供,转染试剂
实施例1
一、提取epc细胞的总rna
使用takara公司的rnaisoplus试剂盒进行epc的总rna提取,具体操作步骤如下:
(1)收集细胞:吸出细胞培养基,用pbs清洗细胞一次;
(2)每10cm2生长的培养细胞加入1-2ml的rnaisoplus试剂,轻轻晃动,确保裂解液均匀分布于细胞表面;
(3)将内含细胞的裂解液转移至离心管中,用移液枪反复吹吸直至裂解液中无明显沉淀;
(4)室温静置5min,然后从核蛋白中分离rna;
(5)向上述步骤(4)的匀浆裂解液中加入氯仿(rnaisoplus的1/5体积量),盖紧离心管盖,混合至溶液乳化呈乳白色;
(6)室温静置5min;;
(7)12,000g,4℃离心15min。从离心机中小心取出离心管,此时匀浆液分为三层,即:无色的上清液(含rna)、中间的白色蛋白层(大部分为dna)及带有颜色的下层有机相;
(8)吸取上清液转移至另一新的离心管中(切勿吸出白色中间层);
(9)向上清中加入0.5-1倍rnaisoplus体积的异丙醇,上下颠倒离心管充分混匀后,置于室温下静置10分钟;
(10)12,000g,4℃离心10min。一般在离心后,试管底部会出现rna沉淀;
(11)小心弃去上清,切勿触及沉淀,残留少量异丙醇没有关系。加入与rnaisoplus等量的75%乙醇,轻轻上下颠倒洗涤离心管管壁,7,500g,4℃离心5min后小心弃去上清,切勿触及沉淀;
(12)打开离心管盖,室温干燥沉淀几分钟。沉淀干燥后,加入适量的rnase-free水溶解沉淀即为epc的总rna;
(13)nanodrop检测所提取rna的浓度及质量,并提供琼脂糖凝胶电泳检测所提取rna的完整性。
二、rna的反转录
以上一步骤所提取的总rna为模板,使用takara公司的试剂盒primescripttm1ststrandcdnasynthesiskit进行反转录,具体流程如下:
(1)按表1(变性反应液配制表)的比例成分在冰上配制反应液的预混液,分装到各反应管中,最后再分别加入模板,将模板与反应液混匀。65℃保温5min后,冰上迅速冷却;
表1
(2)按表2(反转录反应液配置表)的比例成分在冰上配制反转录反应液的预混液,然后取10μl分别添加到上述的各管变性后反应液中;
表2
轻轻混匀后,按照以下条件进行反转录反应:
30℃10min,42℃30min,95℃5min;
然后置于冰上放置,得到epc细胞的cdna,若暂时不用于下一步骤可以存放于-20℃。
三、制备dna模板
(1)使用软件primerpremier5设计各基因的引物,然后在正反向引物的5’端分别加上t7启动子序列,作为完整的引物扩增所研究的基因部分长度片段。本实验添加的t7启动子序列为:5'-taatacgactcactatagggaga-3';
(2)以epc细胞的cdna为模板,用上述所设计的引物,按照表3(制备dna模板反应体系)配制反应液,制备dsrna的dna模板;
表3
(3)先使用基因特异引物(不包含t7启动子序列)的tm值加5℃作为退火温度反应五个循环,再使用完整引物(即基因特异引物加上t7启动子序列)的tm值加5℃作为退火温度循环35个循环进行pcr反应;
(4)琼脂糖凝胶电泳验证产物特异性其目的条带大小,并将扩增产物送公司测序验证其序列。
四、制备目的基因dsrna
(1)第一次使用megascriptrnaikit之前,往2×washsolution加入12mlacs或者更高级别的无水乙醇,即为washingsolution混合均匀后室温保存;
(2)从冰箱取出t7enzymemix直接置于冰上,因为它储存在甘油中,-20℃下不会结冰;
(3)将10×t7reactionbuffer和4种脱氧核苷酸溶液(atp,ctp,gtp,和utp)进行涡旋,一旦解冻立马将4种脱氧核苷酸溶液置于冰上,而10×t7reactionbuffer继续放在室温;
(4)按照表4(体外转录反应体系)在室温下配置反转录反应液,充分混匀反应液,轻弹反应管或者上下吹打然后短暂离心至管子底部,37℃孵育2.5h;
表4
(5)75℃孵育5min,然后自然冷却至室温退火形成dsrna,切勿放在冰上冷却;注:对于≤800nt的dsrna合成反应来说,可不进行退火反应;而对于≥800nt的dsrna合成,则需进行退火反应;
(6)使用te(10mmol/ltris,1mmol/ledta)或者loadingbuffer按1:100比例稀释dsrna,取5μl进行1%琼脂糖凝胶电泳,检测双链形成的完整性及形成效率;
(7)按照表5(核酸酶消化反应体系)在冰上制备rnase消化反应液,然后37℃孵育1h,以除去dna及ssrna。
表5
五、纯化dsrna
(1)按照表6(dsrnabindingmix配方)的配方配制dsrnabindingmix,轻柔地上下吹打混匀;
表6
(2)将上述500μldsrnabindingmix加到超滤管的过滤器上,13,000rpm离心2min,弃滤液,将滤芯放到收集管中;
(3)取500μlwashingsolution到过滤器上,离心弃滤液,重复两次;弃滤液后再空管离心10-30s,以除去残留的液体;
(4)往过滤器加入50μl预热的elutionsolution(≥95℃),然后13,000rpm离心2min;
(5)重复步骤(4),再使用50μlelutionsolution洗脱dsrna到同一收集管中。
(7)测上述产物的rna浓度,然后保存于-20℃;
(7)使用te(10mmol/ltris,1mmol/ledta)或者loadingbuffer按1:5比例稀释dsrna,取5μl进行1%琼脂糖凝胶电泳,检测纯化的dsrna的完整性及双链形成效率。
六、rnaseⅲ消化dsrna
(1)按照表7(rnaseⅲ消化反应体系)配制反应液,混合均匀;
表7
(2)37℃孵育1.5h。
七、搅碎冻融法纯化sirna
将上述dsrna的rnaseⅲ消化产物即sirnamix进行纯化,具体步骤如下。
(1)同时配制大小一致且浓度均为3%的琼脂糖核酸胶,其中一块加入goldview,另外一块不加;
(2)将dsrna的rnaseⅲ消化产物平均分为两份,一份上样于含goldview的核酸胶,另一份上样于不含goldview的核酸胶。注意上样的孔保持一致,两块胶置于同一电泳槽中同时进行电泳;
(3)紫外照射下,含goldview的核酸胶在20bp左右可以看到有明显的条带,此为sirna的目的条带,而不含goldview的核酸胶理论上在同一位置也有相同条带;
(4)将两块胶完全重叠在一起,在紫外灯下切下20bp的目的条带,分别回收到干净的ep管中;
(5)将胶块尽可能切碎,然后置于-80℃冰冻;
(6)将冻结后的胶块取至室温(25℃)解冻融化,加入适量ddh2o,然后使用电动研磨枪进行研磨;
(7)重复步骤(5)、(6)三次,确保胶块完全捣碎;
(8)4℃,12,000rpm离心10min;
(9)用移液枪小心吸取上清转移至干净的离心管中,即为纯化好的sirna。使用nanodrop检测所纯化sirna的浓度,然后计算其摩尔浓度;
(10)琼脂糖凝胶电泳检测纯化效果。
实验结果:
将制备得到的cdna为模板,分别使用设计的含t7启动子序列的完整引物扩增各靶基因含t7启动子序列的dna,用作后续制备dsrna的dna模板,将扩增产物进行琼脂糖凝胶电泳,验证扩增片段的大小及特异性。如图1、图2、图3所示,各基因均扩增出单一明亮的条带,扩增片段大小与预测大小相符,扩增产物测序正确。
rnaseⅲ充分消化dsrna,最终产生的小片段dsrna的介于12-30bp之间。从图4可以看到,8个研究基因的dsrna经rnaseⅲ消化后,片段大小主要为20bp左右,没有看到明显的大片段条带,可见rnaseⅲ消化较为充分,初步获得了各基因的sirna混合物。但是从图中可以看到,rnaseⅲ消化产物中仍有一些dsrna混合物处于20-100bp之间。
为避免酶切后得到的sirnamix中含有的长片段对细胞会产生毒性作用,从中挑取5个sirnamix进行琼脂糖凝胶电泳,然后将20bp左右的条带进行切胶回收,捣碎并且反复冻融,以期sirna从中释放出来,得到不含>30bp片段的sirna。将纯化产物进行琼脂糖凝胶电泳,从图5、图6、图7中可见,未纯化的sirnamix确实存在一定较长片段,而经过纯化后,电泳条带单一而且大小为20bp左右,说明基本上去除了这些大片段,得到纯度较高的sirna。
实施例2
一、sirna转染epc
(1)前一日接种epc细胞:使用含10%fbs不含双抗的mem培养基将epc接种至24孔板,使得次日细胞贴壁后覆盖率为60%-80%。每个实验组设置3个重复孔;
(2)制备sirna-lipid复合体:
①首先使用opti-mem分别稀释rnaimaxreagent和上述制备的sirna,按照适当倍数稀释sirna,脂质体的稀释按照每孔1.5μlrnaimaxreagent+23.5μlopti-mem的比例进行稀释;
②将稀释好的sirna与脂质体按照体积比1:1的比例混合,室温孵5min。因rnaseⅲ消化得到的sirna只要较低的浓度就可以达到化学合成sirna较高浓度得到的效果,结合实验前期探索高浓度25nmol/l、10nmol/l、5nmol/l甚至1nmol/l时基因表达下降率不高,故本研究设置转染时设置较低浓度,每个基因sirna终浓度分别设置0.1nmol/l与0.5nmol/l两个浓度;
实验对照组设置如下:
a)正常细胞对照组(blank组):未经任何转染处理的细胞,用于观测整个实验过程细胞的生长状态及分析的参考;
b)转染试剂对照组(mock组):使用转染试剂进行转染,但是不加入sirna,用于排除转染试剂对细胞可能的影响及分析参考;
c)阴性对照组(nc组):使用锐博生物公司提供的通用型阴性对照sirna进行转染,用于分析目的基因sirna作用的参照;
(3)每个孔加入50μlsirna-lipid复合体,最终转染体系为500μl,将细胞置于25℃、5%co2培养;
(4)收集样品:转染24h后,收集各个孔的细胞及培养液至新的ep管中。
二、rna反转录
将上述收集的样品反复冻融三次,作为反转录的模板,再使用takara公司的试剂盒primescripttm1ststrandcdnasynthesiskit进行反转录,步骤与前面的rna反转录相同。
三、检测各基因沉默效果
使用实时荧光定量pcr(quantitativereal-timepcr,qrt-pcr)的方法检测所研究的八个基因的mrna转录水平的变化。
(1)上述反转录得到的cdna分别稀释100倍,作为qrt-pcr模板,以靶基因设计引物,使用takara公司的tbgreentmpremixextaqtmⅱ(tlirnasehplus)试剂盒进行qrt-pcr。实验时按照表8(qrt-pcr反应体系)的反应体系,冰上配制好试剂预混液,依次分装到384反应板中,最后加cdna模板,每个样品设置三个重复孔。
表8
(2)使用以下反应程序进行荧光定量pcr:
95℃1min
(3)各个反应体系均以β-actin的荧光定量pcr产物为内参照,计算各个目的基因的基因表达相对量,釆用2-△△ct法计算目的基因相对表达量。
实验结果:
将各基因经搅碎冻融法纯化的sirna转染至epc细胞后,以鲤鱼的β-actin基因的表达量作为对照,分析各基因的相对表达量,以检测各sirna对其靶基因的沉默效果。
(1)unigene12835的沉默结果
如图8(a)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的unigene12835sirna转染epc24h后,与nc组相比,两个浓度处理的细胞的unigene12835均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
如图8(b)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的unigene12835sirna转染epc24h后,与mock组相比,两个浓度处理的细胞的unigene12835均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
综上,与nc和mock组相比,unigene12835在rna干扰后表达水平均出现显著下调,其中0.5nmol/l的沉默效果更好,但0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异。
(2)unigene19308的沉默结果
如图9(a)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的unigene19308sirna转染epc24h后,与nc组相比,两个浓度处理的细胞的unigene19308均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
如图9(b)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的unigene19308sirna转染epc24h后,与mock组相比,两个浓度处理的细胞的unigene19308均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
综上,与nc和mock组相比,unigene19308在rna干扰后表达水平均出现显著下调,但0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异。
(3)cl51.contig3的沉默结果
如图10(a)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的cl51.contig3sirna转染epc24h后,与nc组相比,两个浓度处理的细胞的cl51.contig3均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
如图10(b)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的cl51.contig3sirna转染epc24h后,与mock组相比,两个浓度处理的细胞的cl51.contig3均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
综上,与nc和mock组相比,cl51.contig3在rna干扰后表达水平均出现显著下调,但0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异。
(4)unigene13423的沉默效果
如图11(a)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的unigene13423sirna转染epc24h后,与nc组相比,两个浓度处理的细胞的unigene13423均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
如图11(b)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的unigene13423sirna转染epc24h后,与mock组相比,两个浓度处理的细胞的unigene13423均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
综上,与nc和mock组相比,unigene13423在rna干扰后表达水平均出现显著下调,但0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异。
(5)cl3290.contig2的沉默效果
如图12(a)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的cl3290.contig2sirna转染epc24h后,与nc组相比,两个浓度处理的细胞的cl3290.contig2均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
如图12(b)所示,分别使用搅碎冻融法纯化的终浓度0.1nmol/l和0.5nmol/l的cl3290.contig2sirna转染epc24h后,与mock组相比,两个浓度处理的细胞的cl3290.contig2均出现显著性下调(p<0.05),0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异;
综上,与nc和mock组相比,cl3290.contig2在rna干扰后表达水平均出现显著下调,其中0.5nmol/l的沉默效果更好,但0.1nmol/l和0.5nmol/l两个浓度的沉默效果无显著差异。
(6)比较未纯化与纯化的sirna引起的靶基因沉默效果差异
使用终浓度为0.5nmol/l未纯化的cl3290.contig2sirnamix与本申请上述实施例提供的经纯化的cl3290.contig2sirna分别转染epc24h后,如图13(a)所示,与nc组相比,纯化的cl3290.contig2sirna对靶基因沉默效果明显高于未纯化的cl3290.contig2sirnamix;如图13(b)所示,与mock组相比,纯化的cl3290.contig2sirna对靶基因沉默效果也明显高于未纯化的cl3290.contig2sirnamix。
对比例1
将rnaseⅲ消化后的dsrna混合物(步骤参见实施例1)使用30kda的超滤管进行纯化,即:将rnaseⅲ消化后的混合物稀释5倍加载到30kda超滤管中,将超滤管插入配套的微量ep管中,14000g离心20min,收集流出液,理论上即为<30bp的sirna,将超滤管倒扣在一个干净的1.5mlep管中,1000g离心两分钟,收集截留液,理论上即为>30bp的长dsrna。
结果如图14所示,经过30k超滤管纯化,收集的流出液与截留液中均含有<30bp的sirna,截留液中<30bp的sirna反而比流出液中多。并且,从图14可知:unigene13423sirnamix和unigene7122sirnamix经30k超滤管流出样中含有大部分>30bp的sirna,可见30k超滤管截留效果不明显,因此不宜适用于sirna纯化。
综上,本申请实施例在rnaseⅲ消化dsrna获得片段大小不一的混合物的基础上,通过琼脂糖凝胶电泳、切胶后搅碎冻融纯化sirna,与30k超滤管纯化sirna相比,具有以下优点:①操作简便。纯化sirna仅需经过琼脂糖凝胶电泳,胶块研磨冻融等简单步骤,只要具备电泳仪和超低温冰箱的普通分子实验室均可进行,并且无需经过复杂sirnamix稀释以及乙醇浓缩等步骤。②成本低廉。搅碎冻融纯化sirna只需要购买琼脂糖,市售价格约为150元/100g,按照一块琼脂糖凝胶需要0.5g琼脂糖配制,可以上样11个孔进行电泳,则每纯化11个sirna成本为7.5元。30k超滤管市售价格约为1423元/24个,不考虑额外的乙醇浓缩等步骤,通过30k超滤管同样纯化11个sirna成本约为652元,远远高于搅碎冻融纯化sirna。③纯化效果好。搅碎冻融法纯化sirna通过琼脂糖凝胶电泳之后,准确切下<30bp的sirna进行纯化,纯化的sirna绝对不含>30bp的rnaseⅲ消化产物,纯化效果好。而经过30k超滤管纯化sirna,收集的流出液与截留液中均含有<30bp的sirna,并且流出样中含有大部分>30bp的sirna,纯化效果不明显。
以上所述仅为本申请的较佳实施例而已,并不用以限制本申请,凡在本申请的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本申请的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!