一种改进的沙利度胺制备方法与流程
1.本发明属于医药化工领域,具体涉及一种改进的沙利度胺制备方法。
背景技术:
2.沙利度胺(thalidomide)是一种合成的谷氨酸衍生物,其化学名称为(
±
)-n-(2,6-二氧代-3-哌啶基)-邻苯二甲酰亚胺,结构式如下:
[0003][0004]
沙利度胺于1957年首先在德国上市,主要用于催眠、镇静和妇女妊娠早期止吐。但60年代初出现的反应停事件暴露出了沙利度胺具有严重的致畸副作用,随后沙利度胺撤出了医药市场。
[0005]
近年来,科学家对沙利度胺的生物活性进行了深入地研究,特别是在免疫、抗炎、抗血管生成的药理和一些疑难病症上的临床治疗研究中取得了令人欣喜的结果,沙利度胺再次引起关注。1998年美国fda批准celgene公司生产的沙利度胺胶囊用于治疗麻风性结节性红斑(enl)。2006年5月,美国fda又批准celgene公司生产的沙利度胺胶囊用于治疗多发性骨髓瘤(mm)。2010年沙利度胺在中国被批准上市,用于控制瘤型麻风反应症。另外,在临床诊疗指南血液学分册中沙利度胺可以治疗多发性骨髓瘤。在临床诊疗指南风湿学分册中沙利度胺可以治疗强直性脊柱炎和白塞氏症。
[0006]
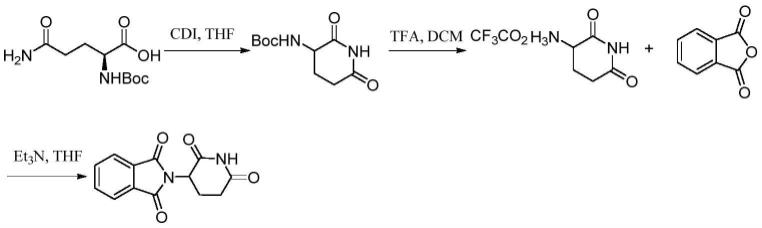
专利wo2005028436公开了一种沙利度胺的合成方法,该方法以boc-l-谷氨酰胺为起始原料,以羰基二咪唑为缩合试剂在四氢呋喃中回流进行关环,然后在二氯甲烷中用三氟乙酸脱boc保护基得到3-氨基-2,6-哌啶二酮三氟乙酸盐,最后以三乙胺为缚酸剂在四氢呋喃中与邻苯二甲酸酐反应得到沙利度胺,反应总收率31%。反应过程如下:
[0007][0008]
专利wo2017081701公开了一种沙利度胺的制备方法,该方法以3-氨基-2,6-哌啶二酮盐酸盐和邻苯二甲酸酐为原料,以三乙胺为缚酸剂,在溶剂(如乙酸、吡啶、二甲基亚砜、n,n-二甲基甲酰胺)中回流反应得到沙利度胺。该方法以化工中间体3-氨基-2,6-哌啶二酮盐酸盐为原料,成本较高;工艺反应温度高,均为高沸点溶剂回流;乙酸做溶剂收率95%,但乙酸气味刺激性大,且后处理产生大量的酸性废液;吡啶做溶剂收率降低至67.7%,且吡啶刺激性大,后处理产生大量的碱性废液;二甲基亚砜做溶剂收率降低至
72%,且回流反应会产生大量刺激性二甲硫醚,污染大气环境;n,n-二甲基甲酰胺做溶剂收率降低至49.7%。反应过程如下:
[0009][0010]
综上所述,本领域迫切需要开发一种操作简便安全,适合工业化大规模生产,生产成本低且纯度高的沙利度胺的制备方法。
技术实现要素:
[0011]
本发明的目的是提供一种操作简便安全,适合工业化大规模生产,生产成本低且纯度高的沙利度胺的制备方法。
[0012]
本发明的第一方面,提供了一种制备沙利度胺的方法,包括以下步骤:
[0013]
(a)提供boc-3-氨基-2,6-哌啶二酮(式iii),将式iii化合物与式iv化合物反应,从而制得沙利度胺
[0014][0015]
在另一优选例中,所述方法可大规模生产。
[0016]
在另一优选例中,所述方法的产量大于100g。
[0017]
在另一优选例中,所述的步骤(a)可以在无催化剂体系中进行。
[0018]
在另一优选例中,所述的步骤(a)在130~160℃的无催化剂体系进行。
[0019]
在另一优选例中,所述的步骤(a)可以在催化剂存在下进行,
[0020]
其中,所述催化剂选自对甲苯磺酸和/或其水合物、氯化氢、硫酸,或其组合。
[0021]
在另一优选例中,所述的步骤(a)在100~130℃的存在催化剂的体系中进行。
[0022]
在另一优选例中,所述的步骤(a)中,催化剂与式iii化合物的质量比为1-5:50。
[0023]
在另一优选例中,所述的步骤(a)中,式iii化合物通过以下步骤制备:
[0024]
(aa1)在水和有机溶剂中,在碱性介质中,l-谷氨酰胺(式i)上保护基得到含有式ii化合物的反应混合物
[0025][0026]
其中,所述的碱性介质为有机碱或有机碱与无机碱的组合;
[0027]
(aa2)用含有式ii化合物的反应混合物与缩合剂进行分子内关环反应,分离后得到aoc-3-氨基-2,6-哌啶二酮(式iii)
[0028][0029]
在另一优选例中,所述的步骤(aa1)中,有机碱选自三乙胺、n,n-二异丙基乙胺、n,n-二异丙基甲胺,或其组合;无机碱选自氢氧化钾、碳酸钾、碳酸氢钾、氢氧化钠、碳酸钠、碳酸氢钠,或其组合。
[0030]
在另一优选例中,所述的有机碱选自三乙胺、n,n-二异丙基乙胺,或其组合。
[0031]
在另一优选例中,所述的碱性介质选自三乙胺、n,n-二异丙基乙胺、三乙胺和氢氧化钾、三乙胺和碳酸钾,或其组合。
[0032]
在另一优选例中,所述的有机碱与l-谷氨酰胺的摩尔比为0.1~2:1;所述的无机碱与l-谷氨酰胺的摩尔比为0~2:1。
[0033]
在另一优选例中,所述的有机碱与l-谷氨酰胺的摩尔比为0.1~1:1;所述的无机碱与l-谷氨酰胺的摩尔比为0.8~1.2:1。
[0034]
在另一优选例中,所述的步骤(aa1)包括步骤:向含式i化合物的原料水溶液中滴加二碳酸二叔丁酯的有机溶剂溶液进行上保护基的反应。
[0035]
在另一优选例中,所述的步骤(aa1)中,水和有机溶剂的体积比为1.5~3:1,更佳地为1.5~2:1。
[0036]
在另一优选例中,所述的步骤(aa1)包括选自下组的后处理:萃取、浓缩、溶剂置换,或其组合。
[0037]
在另一优选例中,所述的步骤(aa2)中,溶剂置换的置换溶剂为1,4-二氧六环、乙二醇、二甲醚,或其组合;优选1,4-二氧六环。
[0038]
在另一优选例中,所述的步骤(aa2)中,缩合剂分批加入。
[0039]
在另一优选例中,所述的步骤(aa2)中,在加入缩合剂之前还加入4-二甲氨基吡啶作为催化剂。
[0040]
在另一优选例中,所述的步骤(a)包括子步骤:
[0041]
(a1)在第一溶剂中,用所述的式iii化合物与邻苯二甲酸酐(式iv)进行反应,从而得到含有沙利度胺(式v)的反应混合物;和
[0042]
(a2)含有沙利度胺(式v)的反应混合物经第二溶剂析晶、分离、精制得到沙利度胺成品。
[0043]
在另一优选例中,所述的第一溶剂选自n-甲基吡咯烷酮(nmp)、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac),或其组合;所述的第二溶剂为水。
[0044]
在另一优选例中,所述的第一溶剂与第二溶剂的体积比为1.5~3:1,更佳地为,1.5~2:1。
[0045]
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
[0046]
图1为实施例3(放大20l批次)中boc-3-氨基-2,6-哌啶二酮的hplc谱图。
[0047]
图2为实施例3(放大20l批次)中沙利度胺粗品的hplc谱图。
[0048]
图3为实施例3(放大20l批次)中沙利度胺成品的hplc谱图。
具体实施方式
[0049]
本发明人通过长期而深入的研究,首次意外地发现,在不使用3-氨基-2,6-哌啶二酮的酸式盐和缚酸剂的合成路径的条件下,不需要脱boc保护基直接使用boc-3-氨基-2,6-哌啶二酮和邻苯二甲酸酐进行反应即可以高收率(60%左右)大批量合成高纯度(99.8%以上)的沙利度胺粗品,进一步精制后得到高纯度(99.98%或更高)的沙利度胺成品。同时本发明人意外地发现在有机碱存在下,可降低boc-l-谷氨酰胺合成工艺中杂质含量,实现了boc-3-氨基-2,6-哌啶二酮的连续生产。与现有工艺相比,本发明所述方法显著缩短了工艺步骤与生产周期,适合大规模工业化生产且在降低生产成本的同时保证高收率和高纯度。基于上述发现,发明人完成了本发明。
[0050]
改进的沙利度胺制备方法
[0051]
本发明提供了一种改进的制备沙利度胺的方法,包括以下步骤:
[0052][0053]
(1)制备boc-3-氨基-2,6-哌啶二酮的步骤,包括步骤:
[0054]
(1.1)在水中,在有机碱或有机碱与无机碱组合物的存在下,加入l-谷氨酰胺(式i),和二碳酸二叔丁酯的四氢呋喃溶液,经反应、后处理及1,4-二氧六环溶剂置换,得到含有boc-l-谷氨酰胺(式ii)的1,4-二氧六环溶液;和
[0055]
(1.2)含有boc-l-谷氨酰胺(式ii)的1,4-二氧六环溶液加入4-二甲氨基吡啶和羰基二咪唑进行分子内关环得到含有boc-3-氨基-2,6-哌啶二酮的反应混合物,然后从boc-3-氨基-2,6-哌啶二酮的反应混合物中分离得到boc-3-氨基-2,6-哌啶二酮(式iii);
[0056]
以及,(2)制备沙利度胺的步骤,包括步骤:
[0057]
(2.1)在第一溶剂中,将boc-3-氨基-2,6-哌啶二酮(式iii)与邻苯二甲酸酐(式iv)在加或不加催化剂的条件下进行反应,从而得到含有沙利度胺(式v)的反应混合物;和
[0058]
(2.2)向含有沙利度胺(式v)的反应混合物加入第二溶剂析晶从而分离得到沙利度胺粗品。沙利度胺粗品经二甲基亚砜和甲醇结晶得到沙利度胺成品。
[0059]
在另一优选例中,所述的步骤(1.1)中,有机碱为三乙胺、n,n-二异丙基乙胺,或其组合物;无机碱为氢氧化钾、碳酸钾、碳酸氢钾、氢氧化钠、碳酸钠、碳酸氢钠,或其组合物;
[0060]
在另一优选例中,所述的步骤(1.1)中,有机碱与l-谷氨酰胺的摩尔比为0.1~
2.0:1.0(较佳地,为0.1~1.0:1.0;更佳地,为0.4:1.0);
[0061]
在另一优选例中,所述的步骤(1.1)中,无机碱与l-谷氨酰胺的摩尔比为0.5~2.0:1.0(较佳地,为0.8~1.2:1.0;更佳地,为1.0:1.0);
[0062]
在另一优选例中,所述的步骤(2.1)可以加或不加催化剂。
[0063]
在另一优选例中,所述的步骤(2.1)中,无催化剂的反应体系的反应温度为130~160℃。
[0064]
在另一优选例中,所述的步骤(2.1)中,存在催化剂的反应体系的反应温度为100~130℃。
[0065]
在另一优选例中,所述步骤(2.1)中,催化剂选自下组:对甲基苯磺酸及其水合物、盐酸、硫酸,或其组合;
[0066]
在另一优选例中,所述的步骤(2.1)中,第一溶剂为n-甲基吡咯烷酮(nmp)、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac),或其组合物;
[0067]
在另一优选例中,所述的步骤(2.2)中,第二溶剂为水。
[0068]
与现有技术相比,本发明的主要优点有:
[0069]
(1)本发明的工艺通过常规方法实现无脱保护剂存在下使用boc-3-氨基-2,6-哌啶二酮直接与邻苯二甲酸酐反应,从而以高产率(总产率在60%左右)制得沙利度胺,该方法显著缩短工艺步骤与生产周期,工艺更安全环保,适合工业化生产;
[0070]
(2)本发明通过在有机碱或有机碱及无机碱的组合物存在下进行上boc保护基,显著降低boc-l-谷氨酰胺合成工艺中杂质焦谷氨酸的含量,经简单后处理即可用于下一步反应,实现了连续反应,工艺操作更简洁,有利于放大生产;
[0071]
(3)本发明的工艺原料成本低廉,可进行大规模生产,且产品质量高,精制后纯度达到99.98%甚至更高,有关物质符合原料药标准。
[0072]
下面结合具体实施,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。
[0073]
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
[0074]
实施例1沙利度胺的制备(小试批次1)
[0075]
实施例1.1 boc-3-氨基-2,6-哌啶二酮的制备(小试批次1)
[0076]
将水600ml加入2l夹套瓶中,加入氢氧化钾57.5g和三乙胺41.5g,搅拌至固体溶解,然后10℃以下加入l-谷氨酰胺150.2g。用恒压滴液漏斗缓慢滴加二碳酸二叔丁酯268.8g的四氢呋喃(300ml)溶液,用时约0.5h。滴加完后,升温至30℃反应。tlc检测反应完毕后加入乙酸乙酯300ml搅拌10分钟,静置分层,分去有机相。水相加入3m稀盐酸(浓盐酸120ml溶解于水360ml)。水相用乙酸乙酯300ml和四氢呋喃300ml混合溶剂萃取3次,合并萃取的有机相。有机相用饱和食盐水600ml洗涤2次。
[0077]
将洗涤后的有机相分批转入2l夹套瓶中,减压浓缩并置换成二氧六环溶液,溶液体积约1.1l,加入4-二甲胺基吡啶5.64g并搅拌溶解。调整温度至25℃以下,分批加入羰基二咪唑180.0g。升温至50℃并保温反应,tlc监测反应结束后停止反应,将反应液减压浓缩
至600ml以下(内温≤55℃)并加水920ml析出固体。抽滤,用水900ml洗滤饼,真空干燥得白色固体boc-3-氨基-2,6-哌啶二酮166.1g,收率70.9%,hplc纯度(面积归一法)99.85%。
[0078]
实施例1.2沙利度胺的制备(小试批次1)
[0079]
将boc-3-氨基-2,6-哌啶二酮157.97g、邻苯二甲酸酐123.02g和n-甲基吡咯烷酮950ml加入2l三颈瓶中,氮气保护,升温至130~160℃保温反应。hplc监测反应完毕后停止反应,反应液降温至60~70℃,滴加475ml纯化水并降温析出固体,抽滤,滤饼用纯化水洗涤滤饼,湿品真空干燥得沙利度胺粗品167.96g,收率94.0%,hplc纯度(面积归一法)99.85%。
[0080]
将沙利度胺粗品165.40g用二甲基亚砜和甲醇重结晶,得到白色固体沙利度胺成品152.75g,收率92.4%,hplc纯度(面积归一法)99.99%。
[0081]
小试批次1中,从原料l-谷氨酰胺制备沙利度胺成品的总收率为61.6%。
[0082]
实施例2沙利度胺的制备(小试批次2)
[0083]
实施例2.1 boc-3-氨基-2,6-哌啶二酮的制备(小试批次2)
[0084]
将水600ml加入2l夹套瓶中,加入氢氧化钾57.5g和三乙胺41.5g,搅拌至固体溶解,然后10℃以下加入l-谷氨酰胺150.0g。用恒压滴液漏斗缓慢滴加二碳酸二叔丁酯268.8g的四氢呋喃(300ml)溶液,用时约0.5h。滴加完后,升温至30℃反应。tlc检测反应完毕后加入乙酸乙酯300ml搅拌10分钟,静置分层,分去有机相。水相加入3m稀盐酸(浓盐酸120ml溶解于水360ml)。水相用乙酸乙酯300ml和四氢呋喃300ml混合溶剂萃取3次,合并萃取的有机相。有机相用饱和食盐水600ml洗涤2次。
[0085]
将洗涤后的有机相分批转入2l夹套瓶中,减压浓缩并置换成二氧六环溶液,溶液体积约1.1l,加入4-二甲胺基吡啶5.64g并搅拌溶解。调整温度至25℃以下,分批加入羰基二咪唑180.0g。升温至50℃并保温反应,tlc监测反应结束后停止反应,将反应液减压浓缩至600ml以下(内温≤55℃)并加水920ml析出固体。抽滤,用水900ml洗滤饼,真空干燥得白色固体boc-3-氨基-2,6-哌啶二酮160.2g,收率68.4%,hplc纯度(面积归一法)98.89%。
[0086]
实施例2.2沙利度胺的制备(小试批次2)
[0087]
将boc-3-氨基-2,6-哌啶二酮157.97g、邻苯二甲酸酐123.02g和n-甲基吡咯烷酮950ml加入2l三颈瓶中,氮气保护,升温至130~160℃保温反应。hplc监测反应完毕后停止反应,反应液降温至60~70℃,滴加475ml纯化水并降温析出固体,抽滤,滤饼用纯化水洗涤滤饼,湿品真空干燥得沙利度胺粗品167.96g,收率94.0%,hplc纯度(面积归一法)99.85%。
[0088]
将沙利度胺粗品165.40g用二甲基亚砜和甲醇重结晶,得到白色固体沙利度胺成品152.75g,收率92.4%,hplc纯度(面积归一法)99.99%。
[0089]
小试批次2中,从原料l-谷氨酰胺制备沙利度胺成品的总收率为59.4%。
[0090]
实施例3沙利度胺的制备(放大20l批次)
[0091]
实施例3.1 boc-3-氨基-2,6-哌啶二酮的制备(放大20l批次)
[0092]
将水6.0l加入2l夹套瓶中,加入氢氧化钾576.2g和三乙胺415.3g,搅拌至固体溶解,然后10℃以下加入l-谷氨酰胺1500.2g。用恒压滴液漏斗缓慢滴加二碳酸二叔丁酯2689.1g的四氢呋喃(3.0l)溶液,用时约0.5h。滴加完后,升温至30℃反应。tlc检测反应完毕后加入乙酸乙酯3.0l搅拌10分钟,静置分层,分去有机相。水相加入3m稀盐酸(浓盐酸
1.2l溶解于水3.6l)。水相用乙酸乙酯3.0l和四氢呋喃3.0l混合溶剂萃取3次,合并萃取的有机相。有机相用饱和食盐水6.0l洗涤2次。
[0093]
将洗涤后的有机相分批转入20l玻璃反应釜中,减压浓缩并置换成二氧六环溶液,溶液体积约11l,加入4-二甲胺基吡啶56.4g并搅拌溶解。调整温度至25℃以下,分批加入羰基二咪唑1799.9g。升温至50℃并保温反应,tlc监测反应结束后停止反应,将反应液减压浓缩至6.0l以下(内温≤55℃)并加水9.2l析出固体。抽滤,用水9.0l洗滤饼,真空干燥得白色固体boc-3-氨基-2,6-哌啶二酮1530.7g,收率65.3%,hplc纯度(面积归一法)99.90%。hplc谱图如图1所示。
[0094]
实施例3.2沙利度胺的制备(放大20l批次)
[0095]
将boc-3-氨基-2,6-哌啶二酮1528.7g、邻苯二甲酸酐1190.4g和n-甲基吡咯烷酮9.2l加入20l玻璃反应釜中,氮气保护,升温至140~160℃保温反应。hplc监测反应完毕后停止反应,反应液降温至60~70℃,滴加4.6l纯化水并降温析出固体,抽滤,滤饼用纯化水洗涤滤饼,湿品真空干燥得沙利度胺粗品1685.0g,收率97.4%,hplc纯度(面积归一法)99.96%。沙利度胺粗品的hplc谱图如图2所示。
[0096]
将沙利度胺粗品1661.0g用二甲基亚砜和甲醇重结晶,得到白色固体沙利度胺成品1539.2g,收率92.7%,hplc纯度(面积归一法)99.99%。沙利度胺精品的hplc谱图如图2所示。
[0097]
放大20l批次中,从原料l-谷氨酰胺制备沙利度胺成品的总收率为59.0%。
[0098]
实施例4沙利度胺粗品的制备
[0099]
将boc-3-氨基-2,6-哌啶二酮20.00g、邻苯二甲酸酐15.69g、对甲苯磺酸一水合物1.75g和n-甲基吡咯烷酮120ml加入反应瓶中,氮气保护,升温至120℃保温反应。hplc监测反应完毕后停止反应,反应液降温至60~70℃,滴加60ml纯化水并降温析出固体,抽滤,滤饼用纯化水洗涤滤饼,湿品真空干燥得沙利度胺粗品20.01g,收率88.4%,hplc纯度(面积归一法)99.88%。
[0100]
实施例5沙利度胺粗品的制备
[0101]
将boc-3-氨基-2,6-哌啶二酮20.00g、邻苯二甲酸酐15.58g、对甲苯磺酸一水合物1.72g和n,n-二甲基乙酰胺120ml加入反应瓶中,氮气保护,升温至130℃保温反应。hplc监测反应完毕后停止反应,反应液降温至60~70℃,滴加60ml纯化水并降温析出固体,抽滤,滤饼用纯化水洗涤滤饼,湿品真空干燥得沙利度胺粗品21.13g,收率93.4%,hplc纯度(面积归一法)99.96%。
[0102]
实施例6沙利度胺粗品的制备
[0103]
将boc-3-氨基-2,6-哌啶二酮20.00g、邻苯二甲酸酐15.58g、氯化氢0.64g(氯化氢二氧六环溶液,浓度4.0mol/l,用量4.4ml)和n-甲基吡咯烷酮120ml加入反应瓶中,氮气保护,升温至130℃保温反应。hplc监测反应完毕后停止反应,反应液降温至60~70℃,滴加60ml纯化水并降温析出固体,抽滤,滤饼用纯化水洗涤滤饼,湿品真空干燥得沙利度胺粗品21.97g,收率97.1%,hplc纯度(面积归一法)99.88%。
[0104]
实施例7沙利度胺粗品的制备
[0105]
将boc-3-氨基-2,6-哌啶二酮20.02g、邻苯二甲酸酐15.58g、硫酸0.885g和n-甲基吡咯烷酮120ml加入反应瓶中,氮气保护,升温至130℃保温反应。hplc监测反应完毕后停止
反应,反应液降温至60~70℃,滴加60ml纯化水并降温析出固体,抽滤,滤饼用纯化水洗涤滤饼,湿品真空干燥得沙利度胺粗品20.98g,收率92.7%,hplc纯度(面积归一法)99.96%。
[0106]
实施例1-7表明在由boc-3-氨基-2,6-哌啶二酮制备沙利度胺的步骤中,使用酸性催化剂可以降低反应温度。
[0107]
另外,对boc-l-谷氨酰胺的合成工艺中使用无机碱和有机碱对产物纯度、主要杂质焦谷氨酸含量及反应收率的影响进行了考察,结果如下表所示:
[0108]
表1:无机碱和有机碱对boc-l-谷氨酰胺的合成工艺的影响
[0109][0110]
*1:产品纯度和焦谷氨酸含量是把反应液直接送检hplc测得,数值采用面积归一法。
[0111]
*2:反应收率是把反应液直接送检,由hplc外标法测得产品含量,再结合反应液重量计算而来。
[0112]
上述实验数据表明:单独采用无机碱,会造成部分l-谷氨酰胺转化为焦谷氨酸,从而造成产物纯度和反应收率下降;而采用有机碱或有机碱与无机碱联用可以有效避免焦谷氨酸的产生,反应液纯度可以达到97%以上,经简单后处理后即可用于下一步反应。
[0113]
讨论
[0114]
本发明在有机碱或有机碱与无机碱组合存在下进行boc保护基反应,可显著降低反应副产物焦谷氨酸的产生,从而使所得的反应混合物经简单处理后即可用于下一步反应,实现boc-3-氨基-2,6-哌啶二酮的连续生产。
[0115]
本发明在不使用脱保护剂,预先将boc-3-氨基-2,6-哌啶二酮脱保护的条件下,直
接将boc-3-氨基-2,6-哌啶二酮与邻苯二甲酸酐进行常规反应,从而得到沙利度胺,且不使用相对严苛的无法进行大规模生产的方法(如微波法),显著缩短了工艺步骤和工艺周期,且总收率较高。制得的沙利度胺纯度高(99.98%或以上),粗品纯度可达99.8%。
[0116]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1