一种苯并噁嗪-4-酮衍生物的制备方法与流程
一种苯并噁嗪
‑4‑
酮衍生物的制备方法
技术领域
1.本发明涉及一种苯并噁嗪
‑4‑
酮衍生物的制备方法,具体涉及一种2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑
4h
‑
3,1
‑
苯并噁嗪
‑4‑
酮衍生物的制备方法,属于化学合成技术领域。
背景技术:
[0002]2‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑
4h
‑
3,1
‑
苯并噁嗪
‑4‑
酮衍生物是合成双酰胺类农药杀虫剂的重要中间体,例如2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑8‑
甲基4h
‑
3,1
‑
苯并恶嗪
‑4‑
酮是合成氯虫苯甲酰胺的前体。
[0003]
cn1678192a中给出了两种这类苯并噁嗪
‑4‑
酮衍生物的合成方法,一是在叔胺存在下,由甲基磺酰氯,式(2)对应的吡唑甲酸,以及式(1)的邻氨基苯甲酸反应制备;二是由相应结构的靛红酸酐和式(2)所示的吡唑酰基氯在吡啶或吡啶乙腈溶剂中反应制备。
[0004]
以上两种方法经我们研发发现,存在收率偏低,有机碱用量过大产生大量三废难处理,不适合工业化生产的问题。
[0005]
因此,需要另行开发出一种成本较低,安全高效合成方法合成这类苯并噁嗪
‑4‑
酮衍生物。
技术实现要素:
[0006]
本发明所要解决的技术问题是针对现有技术中存在的不足,而提供一种如式3所示的苯并噁嗪
‑4‑
酮衍生物(2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑
4h
‑
3,1
‑
苯并恶嗪
‑4‑
酮衍生物)的制备方法,本发明方法中,以2
‑
氨基
‑5‑
氯
‑
苯甲酸衍生物和3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
羰基氯作为原料直接进行环化从而得到目标产物,合成收率高,三废产生量小。
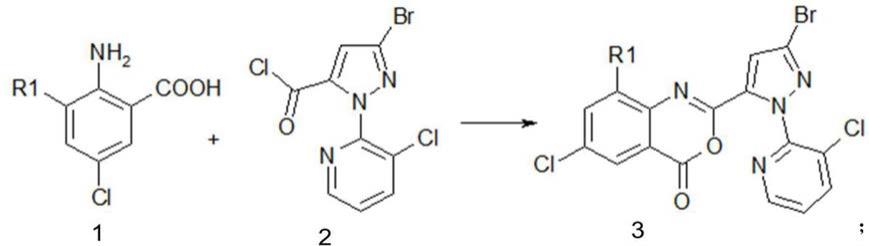
[0007]
为了实现上述目的,本发明采用如下技术方案:
[0008]
一种苯并噁嗪
‑4‑
酮衍生物的制备方法,包括以下步骤:
[0009]
在有机溶剂中,以式1所示的2
‑
氨基
‑5‑
氯
‑3‑
取代的苯甲酸和式2所示的3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
羰基氯作为原料,向体系中加入碱然后在低温条件下滴加磺酰氯溶液,式1所示化合物和式2所示化合物升温后进行环化反应,环化反应结束后,经降温、过滤、洗涤、烘干后得到苯并噁嗪
‑4‑
酮衍生物,即式3所示的2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑
4h
‑
3,1
‑
苯并恶嗪
‑4‑
酮;化学反应式如下所示:
[0010][0011]
其中:r1为me或cl。
[0012]
上述技术方案中,所述的有机溶剂,为乙腈、丙酮、甲基乙基酮、乙酸乙酯、氯仿、二氯乙烷、甲苯中的任意一种、两种及以上的混合物,优选为乙腈。
[0013]
上述技术方案中,所述的式2所示的化合物与有机溶剂的质量比为1:1.5~3,优选为1:2。
[0014]
上述技术方案中,所述的式1所示化合物和式2所示化合物,摩尔比为1:0.8~1,优选为1:0.95。
[0015]
上述技术方案中,所述的碱为吡啶、3
‑
甲基吡啶、2
‑
甲基吡啶中的任意一种,优选为3
‑
甲基吡啶。
[0016]
上述技术方案中,所述的碱,与式2所示化合物的摩尔比为1.5~4:1,优选为2.2:1。
[0017]
上述技术方案中,所述的磺酰氯溶液,指的是将磺酰氯溶于溶剂后得到的溶液;所述的磺酰氯为甲磺酰氯、丙磺酰氯、苯磺酰氯中的任意一种,优选为甲磺酰氯;所述的溶剂为氯仿、二氯乙烷、二氯甲烷、乙腈、乙酸乙酯中的任意一种、两种及以上的混合物,优选为乙腈。
[0018]
上述技术方案中,所述的磺酰氯溶液,其中的溶质磺酰氯,与式2所示化合物的的摩尔比为1~5:1,优选为1.2:1。
[0019]
上述技术方案中,滴加磺酰氯溶液的温度为
‑
20℃~15℃,优选为
‑
5℃~10℃。
[0020]
上述技术方案中,所述的环化反应,反应的温度为5℃~35℃,优选为25℃,反应时间为2h~6h,优选为2h。
[0021]
上述技术方案中,所述的降温,指的是降至0℃~10℃,优选为5℃。
[0022]
本发明的技术效果:本发明的2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑
4h
‑
3,1
‑
苯并恶嗪
‑4‑
酮的新的制备方法,操作简单、收率高、成本低、三废少,适合于工业化生产。
具体实施方式
[0023]
以下对本发明技术方案的具体实施方式详细描述,但本发明并不限于以下描述内容:
[0024]
下面结合具体的实施例,对本发明进行阐述:
[0025]
实施例1
[0026]
一种2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑
4h
‑
3,1
‑
苯并恶嗪
‑4‑
酮的制备方法,包括以下步骤:
[0027]
将10.5g(56.7mmol)2
‑
氨基
‑5‑
氯
‑3‑
甲基苯甲酸与17.3g(53.9mmol)3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
羰基氯置于烧瓶中,加入30g乙腈,再加入10g 3
‑
甲基吡啶,降温至0~5℃,滴加含有7.4g甲磺酰氯的乙腈()溶液10g,滴加结束之后,升至25℃搅拌2h,之后将温度降至0~5℃,搅拌0.5h,过滤,滤饼用冰乙腈洗涤,烘干得目标产物23.1g,产率95%。
[0028]
实施例2
[0029]
一种2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑
4h
‑
3,1
‑
苯并恶嗪
‑4‑
酮的制备方法,包括以下步骤:
[0030]
将10.5g(56.7mmol)2
‑
氨基
‑5‑
氯
‑3‑
甲基苯甲酸与17.3g(53.9mmol)3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
羰基氯置于烧瓶中,加入30g乙腈,再加入10g 3
‑
甲基吡啶,降温至0~5℃,滴加含有7.4g甲磺酰氯的乙腈()溶液10g,滴加结束之后,升至25℃搅拌2h,之后将温度降至0~5℃,搅拌0.5h,过滤,滤饼用冰乙腈洗涤,烘干得目标产物22.6g,产率93%。
[0031]
实施例3
[0032]
一种2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑
4h
‑
3,1
‑
苯并恶嗪
‑4‑
酮的制备方法,包括以下步骤:
[0033]
将10.5g(56.7mmol)2
‑
氨基
‑5‑
氯
‑3‑
甲基苯甲酸与17.3g(53.9mmol)3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
羰基氯置于烧瓶中,加入30g乙腈,再加入10g 3
‑
甲基吡啶,降温至0~5℃,滴加含有7.4g甲磺酰氯的乙腈()溶液10g,滴加结束之后,升至25℃搅拌2h,之后将温度降至0~5℃,搅拌0.5h,过滤,滤饼用冰乙腈洗涤,烘干得目标产物23.1g,产率95%。
[0034]
实施例4
[0035]
一种2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑6‑
氯
‑
4h
‑
3,1
‑
苯并恶嗪
‑4‑
酮的制备方法,包括以下步骤:
[0036]
将10.5g(56.7mmol)2
‑
氨基
‑5‑
氯
‑3‑
甲基苯甲酸与17.3g(53.9mmol)3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
羰基氯置于烧瓶中,加入30g乙腈,再加入10g 3
‑
甲基吡啶,降温至0~5℃,滴加含有7.4g甲磺酰氯的乙腈()溶液10g,滴加结束之后,升至25℃搅拌2h,之后将温度降至0~5℃,搅拌0.5h,过滤,滤饼用冰乙腈洗涤,烘干得目标产物22.9g,产率94%。
[0037]
实施例5
[0038]
一种2
‑
[3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
基]
‑
6,8
‑
二氯
‑
4h
‑
3,1
‑
苯并恶嗪
‑4‑
酮的制备方法,包括以下步骤:
[0039]
将11.7g(56.7mmol)2
‑
氨基
‑
3,5
‑
二氯苯甲酸与17.3g(53.9mmol)3
‑
溴
‑1‑
(3
‑
氯
‑2‑
吡啶基)
‑
1h
‑
吡唑
‑5‑
羰基氯置于烧瓶中,加入30g乙腈,再加入10g 3
‑
甲基吡啶,降温至0~5℃,滴加含有7.4g甲磺酰氯的乙腈溶液10g,滴加结束之后,升至25℃搅拌2h,之后将温度降至0~5℃,搅拌0.5h,过滤,滤饼用冰乙腈洗涤,烘干得目标产物23.4g,产率92%。
[0040]
上述实例只是为说明本发明的技术构思以及技术特点,并不能以此限制本发明的保护范围。凡根据本发明的实质所做的等效变换或修饰,都应该涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1