一种淫羊藿的磷酸酯类衍生物及其制备方法和应用
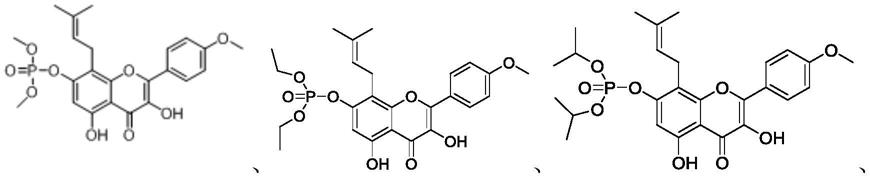
1.本发明属于药物技术领域,更具体地,涉及一种淫羊藿的磷酸酯类衍生物及其制备方法和应用。
背景技术:
2.淫羊藿为我国传统的补益中草药,又名仙灵脾,小檗科淫羊藿属。淫羊藿具有补肾壮阳、强筋健骨、祛风除湿等功效,是一味中医治疗骨质疏松方剂的传统中药材,对其使用均为在中医理论指导下使用淫羊藿植株煎汤、浸酒、熬膏和制丸散等。目前,现代医学针对骨质疏松的治疗主要采用双磷酸盐、降钙素、氟化物和激素替代疗法等,但疗效均存在一定争议,且副作用明显,限制了药物的使用。我国中医药治疗骨质疏松症的历史悠久,对骨质疏松的症状具有明显的改善作用,传统中药价格低廉和副作用低等优点。淫羊藿的主要成分淫羊藿苷具有抗肿瘤功效、壮骨、调节免疫功能、改善心血管功能、抗衰老和抗氧化功能、增强抗毒病能力和增强性功能等生理活性。天然产物作为化学药物本身或药物研发的先导化合物是多年来药物研发人员从事化学药物研发的重要研究方法之一,黄酮苷类化合物淫羊藿苷也成为研究的热点。
3.磷是生物体中不可缺少的元素之一,在体内主要以羟基磷灰石(3ca3(po4)2‑
ca(oh)2)的无机盐形式存在于骨和牙齿中,占其总量的87.6%,部分磷以hpo
42
‑
和h2po4‑
的形式存在于血、尿和组织液中,起到调节ph和ca、p平衡的作用。核酸中极其重要的磷酸双酯,其中磷的含量约9%。作为有机磷小分子化合物,磷酰胺及其酯类衍生物大多具有广泛的的生物活性,它们参与生命有机体内的物质转化并在其生命过程中发货着重要作用,在医药上对一些官能团进行结构改造,以改变其作用方式和生物活性,这对于更好的认识生命体及人类自身,促进新药开发,都具有重要意义。
技术实现要素:
4.为了解决上述现有技术中存在的问题。本发明的目的在于提供一种淫羊藿的磷酸酯类衍生物,该类化合物具有作为抗肿瘤和抗骨质疏松的作用。
5.本发明的另一目的在于提供上述淫羊藿的磷酸酯类衍生物的制备方法。
6.本发明的再一目的在于提供上述淫羊藿的磷酸酯类衍生物的应用。
7.本发明的目的通过下述技术方案来实现:
8.一种淫羊藿的磷酸酯类衍生物,所述的淫羊藿的磷酸酯类衍生物的化学结构式如式(1)所示:
9.10.其中,r1选自选自r2选自h或r1,r3选自h
11.优选地,当r2和r3同时为h时,所述的淫羊藿的磷酸酯类衍生物的分子结构式为:
[0012][0013]
更为优选地,所述的淫羊藿的磷酸酯类衍生物的分子结构式为:
[0014][0014][0015]
优选地,当r2为h且r3为时,所述的淫羊藿的磷酸酯类衍生物的分子结构式为:
[0016]
[0017]
优选地,当r2为r1且r3为时,所述的淫羊藿的磷酸酯类衍生物的分子结构式为:
[0018][0019]
所述的淫羊藿的磷酸酯类衍生物的制备方法,包括如下方法:
[0020]
将淫羊藿素或淫羊藿次苷ⅱ和催化剂加入有机溶剂a中,冰水浴中降温至0℃,溶解后滴入含亚磷酸二甲酯和有机溶剂b的磷酸酯化供体,在20~40℃反应5~12h,反应结束后减压浓缩,用硅胶柱层析方法纯化,得到淫羊藿的磷酸酯类衍生物。
[0021]
优选地,所述的催化剂为三乙胺、哌啶、4
‑
二甲氨基吡啶或二异丙基乙胺中的一种以上;所述的有机溶剂a为乙腈、二甲基亚砜、甲苯、乙醇、四氢呋喃中的一种以上;所述的有机溶剂b为四氯化碳、三氯化碳、三氯溴甲烷或三氯碘甲烷中的一种以上。
[0022]
优选地,所述的淫羊藿素或淫羊藿次苷ⅱ与催化剂的摩尔比为1:(1~6);所述的淫羊藿素或淫羊藿次苷ⅱ与磷酸酯化供体中亚磷酸二甲酯的摩尔比为1:(1~4);所述的淫羊藿素或淫羊藿次苷ⅱ的质量和有机溶剂a的体积比为1g:(100~200)ml;所述的磷酸酯化供体中亚磷酸二乙酯和有机溶剂b的体积比为1:(100~200)。
[0023]
所述的淫羊藿的磷酸酯类衍生物在制备前列腺癌细胞的药物领域中的应用。
[0024]
所述的淫羊藿的磷酸酯类衍生物在制备抗骨质疏松的保健品和药物辅料和药品领域中的应用。
[0025]
本发明的系列淫羊藿的磷酸酯类衍生物的生物实验,验证了淫羊藿次苷ⅱ磷酸酯类衍生物具壮骨的用途。根据药物拼接原理,通过atherton
–
todd反应将磷酸酯基拼接到淫羊藿素和淫羊藿次苷ⅱ中。由淫羊藿素和淫羊藿次苷ⅱ得到淫羊藿类化合物磷酸酯化产物的方法以及此类化合物的药物组合物。淫羊藿次苷ⅱ磷酸酯衍生物及其药物组合物能够促进成骨细胞的钙结节形成率。
[0026]
与现有技术相比,本发明具有以下有益效果:
[0027]
1.本发明的淫羊藿的磷酸酯类衍生物在raw264.7细胞的mtt实验中显示,淫羊藿素磷酸二甲酯和淫羊藿次苷ⅱ磷酸二甲酯的细胞毒性较淫羊藿素和淫羊藿次苷ⅱ显著降低。
[0028]
2.本发明的淫羊藿的磷酸酯类衍生物,在pc
‑
3(前列腺癌细胞)细胞毒实验中显示,部分修饰后衍生物较修饰前细胞毒性显著增强,在同等浓度下可以用于制备预防或治疗癌症的药物。
[0029]
3.本发明的淫羊藿的磷酸酯类衍生物在较低的浓度下(62.5~500nmol/l)就能促
进成骨分化形成钙结节,在显微镜下能够较明显的看出染成红色的钙结节,能够作为治疗骨质疏松的潜在前景。
[0030]
4.本发明的淫羊藿的磷酸酯类衍生物的制备过程简单、成本低廉,结构稳定,便于储存。
附图说明
[0031]
图1实施例1
‑
4中淫羊藿素的磷酸酯化衍生物对成骨细胞矿化结节的影响。
具体实施方式
[0032]
下面结合具体实施例进一步说明本发明的内容,但不应理解为对本发明的限制。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
[0033]
实施例1
[0034]
3,5,7
‑
三羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)(淫羊藿素),淫羊藿素的结构如下所示:
[0035][0036]
由于淫羊藿素存在于淫羊藿中较少,淫羊藿素是从中药淫羊藿中提取分离得到,主要是将淫羊藿苷水解后获得。淫羊藿素是以淫羊藿苷为原料,以固定化α
‑
l
‑
鼠李糖苷酶和葡萄糖苷酶进行水解。取淫羊藿苷5g于250ml圆底烧瓶中加入去离子水50ml,置于60℃的恒温水浴振荡器中,调节ph至9~10,待底物溶解后加入固定化α
‑
l
‑
鼠李糖苷酶1g和葡萄糖苷酶1g,以振荡频率为120r/min反应3h,tlc监控,原料反应完全后,调节ph至3~4,滤液置冰箱至析出,过滤得到呈黄色粉末状晶体的淫羊藿素,具体反应如式(1)所示:
[0037][0038]
所得淫羊藿素的核磁共振光谱为:1h nmr(400mhz,dmso)δ12.39(d,j=13.9hz,1h),10.73(s,1h),9.47(s,1h),8.08(dd,j=37.9,8.9hz,2h),7.07(t,j=40.1hz,2h),6.30(s,1h),5.18(t,j=6.7hz,1h),3.85(s,3h),3.41(t,j=13.8hz,2h),1.75(s,3h),1.55(d,j=63.2hz,3h)。
[0039]
实施例2
[0040]
3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)
磷酸二甲酯的结构式为:
[0041][0042]
将实施例1合成的3,5,7
‑
三羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)(淫羊藿素)(193mg,0.5mmol)溶于20ml四氢呋喃,加入催化剂二异丙基乙胺(dipea,0.348ml,2mmol)和4
‑
二甲氨基吡啶(dmap,6mg,0.05mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二甲酯(46ul,0.5mmol)的ccl4(2ml)中,缓慢升至20℃,薄层色谱(甲醇:二氯甲烷=1:10)监控反应。反应完全后减压浓缩,用柱层析方法(硅胶柱提纯)纯化,得到目标化合物3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二甲酯,其具体反应如式(2)所示:
[0043][0044]
3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二甲酯为淡黄色固体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ11.75(s,1h),8.18(d,j=7.8hz,2h),7.05(d,j=7.9hz,2h),6.88(s,1h),6.60(s,1h),5.21(s,1h),4.10
–
3.76(m,9h),3.62(d,j=6.2hz,2h),1.80(d,j=21.1hz,3h),1.69(s,3h).
31
p nmr(162mhz,cdcl3)δ
‑
4.72(s)。
[0045]
实施例3
[0046]
3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二乙酯的结构式为:
[0047][0048]
将实施例1合成的3,5,7
‑
三羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)(淫羊藿素)(193mg,0.5mmol)溶于四氢呋喃(20ml),加入催化剂二异丙基乙胺(dipea,0.348ml,2mmol)和4
‑
二甲氨基吡啶(dmap,6mg,0.05mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二乙酯(64ul,0.5mmol)的ccl4(2ml)的溶液,缓慢升至20℃,薄层色谱(甲醇:二氯甲烷=1:10)监控反应。反应完全后减压浓缩,柱层析方法(硅胶柱提纯)纯化,得到目标化合物3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡
喃
‑4‑
酮)磷酸二乙酯,具体反应如式(3)所示:
[0049][0050]
3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二乙酯为淡黄色固体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ11.74(s,1h),8.19(d,j=8.8hz,2h),7.05(d,j=8.9hz,2h),6.91(s,1h),6.61(s,1h),5.21(s,1h),4.36
–
4.12(m,4h),3.90(s,3h),3.61(d,j=6.5hz,2h),1.82(s,3h),1.67(d,j=14.3hz,3h),1.38(t,j=7.1hz,6h).
31
p nmr(162mhz,cdcl3)δ
‑
7.02(s)。
[0051]
实施例4
[0052]
3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二异丙酯的结构式为:
[0053][0054]
将实施例1合成的3,5,7
‑
三羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)(淫羊藿素)(0.193g,0.5mmol)溶于四氢呋喃(20ml),加入催化剂二异丙基乙胺(dipea,0.348ml,2mmol)和4
‑
二甲氨基吡啶(dmap,6mg,0.05mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二异丙酯(83ul,0.5mmol)的ccl4(2ml)的溶液,缓慢升至20℃,薄层色谱(甲醇:二氯甲烷=1:10)监控反应。反应完全后减压浓缩,柱层析方法(硅胶柱提纯)纯化,得到目标化合物3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二异丙酯,具体反应如式(4)所示:
[0055][0056]
3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二异丙酯为黄色固体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ11.72(s,1h),8.19(d,j=7.5hz,2h),7.05(d,j=7.5hz,2h),6.95(s,1h),6.61(s,1h),5.21(s,1h),4.80(td,j=12.4,6.2hz,2h),3.90(d,j=1.4hz,3h),3.61(d,j=6.2hz,2h),1.78(d,j=30.8hz,3h),1.66(d,j=23.5hz,3h),1.49
–
1.26(m,12h).
31
p nmr(162mhz,cdcl3)δ
‑
8.77(s)。
[0057]
实施例5
[0058]
3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二异丁酯的结构式为:
[0059][0060]
将实施例1合成的3,5,7
‑
三羟基
‑2‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)(淫羊藿素)(0.193g,0.5mmol,1.0eq)溶于四氢呋喃(20ml),加入催化剂二异丙基乙胺(dipea,0.348ml,2mmol)和4
‑
二甲氨基吡啶(dmap,6mg,0.05mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二异丁酯(99ul,0.5mmol)的ccl4(2ml)的溶液,缓慢升至20℃,薄层色谱(甲醇:二氯甲烷=1:10)监控反应。反应完全后减压浓缩,柱层析方法(硅胶柱提纯)纯化,得到目标化合物3,5
‑
二羟基
‑8‑
(3
‑
甲基
‑2‑
丁烯基)
‑2‑
(4
‑
甲氧基苯基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二异丁酯,具体反应如式(5)所示:
[0061][0062]
3,5
‑
二羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基
‑2‑
烯
‑1‑
基)
‑7‑
(4h
‑
苯并吡喃
‑4‑
酮)磷酸二异丁酯为黄色固体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ11.72(s,1h),8.19(d,j=9.0hz,2h),7.05(d,j=9.0hz,2h),6.92(s,1h),6.61(s,1h),5.21(s,1h),3.95(tt,j=9.6,4.9hz,4h),3.90(d,j=5.7hz,3h),3.62(d,j=6.6hz,2h),2.00(dt,j=13.3,6.7hz,2h),1.82(s,3h),1.69(s,3h),0.92(m,12h),
31
p nmr(162mhz,cdcl3)δ
‑
6.84(s)。
[0063]
实施例6
[0064]
5,7
‑
二羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮(淫羊藿次苷ⅱ)的结构式为:
[0065][0066]
淫羊藿次苷ⅱ的制备方法是以淫羊藿苷为原料,用葡萄糖苷酶进行水解。取淫羊藿苷5g于250ml圆底烧瓶中加入去离子水50ml,置于60℃的恒温水浴振荡器中,调节ph至9~10,待底物溶解后加入葡萄糖苷酶1g,以振荡频率为120r/min反应3h,tlc监控,原料反应完全后,调节ph至3~4,滤液置冰箱至析出,过滤得到呈黄色固体的淫羊藿次苷ii,具体反
应如式(6)所示:
[0067][0068]
淫羊藿次苷ⅱ其核磁共振光谱为:1h nmr(400mhz,dmso)δ12.58
–
12.35(m,1h),9.61(s,1h),8.24
–
7.84(m,2h),7.14(dd,j=8.9,3.5hz,2h),6.63(d,j=11.9hz,1h),5.35(s,1h),5.21(t,j=6.9hz,1h),5.17
–
5.09(m,1h),5.06(d,j=5.1hz,1h),5.03
–
4.97(m,1h),4.64(t,j=5.4hz,1h),3.86(s,3h),3.73(dd,j=10.8,4.8hz,1h),3.48
–
3.40(m,3h),3.32(d,j=3.3hz,2h),1.87
–
1.69(m,3h),1.62(d,j=9.6hz,3h),1.25(s,3h)。
[0069]
实施例7
[0070]5‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二甲酯的结构式为:
[0071][0072]
将实施例6合成的淫羊藿次苷ⅱ(103mg,0.2mmol)溶于乙醇(20ml),加入二催化剂异丙基乙胺(dipea,0.139ml,0.8mmol)和4
‑
二甲氨基吡啶(dmap,2.5mg,0.02mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二甲酯(18ul,0.2mmol)的ccl4(2ml)的溶液,缓慢升至20℃,薄层色谱(甲醇:二氯甲烷=1:5)监控反应。反应完全后减压浓缩,柱层析方法(硅胶柱提纯)纯化,得到目标化合物5
‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二甲酯;其具体反应如式(7)所示:
[0073][0074]5‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二甲酯为黄色固体,其核
磁共振光谱为:1h nmr(400mhz,cdcl3)δ12.38(s,1h),7.78(s,2h),6.98(d,j=7.1hz,2h),6.80(s,1h),5.46(s,1h),5.13(s,1h),4.46(s,1h),4.03
–
3.70(m,12h),3.54(d,j=51.7hz,4h),3.31(s,2h),2.09(s,4h),1.70(d,j=12.1hz,3h),1.64(d,j=19.7hz,3h),1.26(s,3h);
31
p nmr(162mhz,cdcl3)δ
‑
4.84(s)。
[0075]
实施例8
[0076]5‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二乙酯的结构式为:
[0077][0078]
将实施例6合成的淫羊藿次苷ⅱ(103mg,0.2mmol)溶于乙醇(20ml),加入二催化剂异丙基乙胺(dipea,0.139ml,0.8mmol)和4
‑
二甲氨基吡啶(dmap,2.5mg,0.02mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二乙酯(25ul,0.2mmol)的ccl4(2ml)的溶液,缓慢升至20℃,薄层色谱(甲醇:二氯甲烷=1:5)监控反应。反应完全后,减压浓缩,柱层析方法(硅胶柱提纯)纯化,得到目标化合物,其具体反应如式(8)所示:
[0079][0080]5‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二乙酯为黄色固体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ12.34(s,1h),7.79(d,j=8.2hz,2h),6.97(d,j=6.8hz,2h),6.83(s,1h),5.43(d,j=11.2hz,1h),5.13(d,j=5.8hz,1h),4.60
–
4.29(m,3h),4.27
–
4.19(m,4h),3.81(s,3h),3.68
–
3.43(m,3h),3.31(s,1h),1.68(d,j=20.3hz,6h),1.36(t,j=7.0hz,6h),1.26(s,3h);
31
p nmr(162mhz,cdcl3)δ
‑
7.19(s)。
[0081]
实施例9
[0082]5‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二丙酯的结构式为:
[0083][0084]
将实施例6合成的淫羊藿次苷ⅱ(103mg,0.2mmol)溶于乙醇(20ml),加入二催化剂异丙基乙胺(dipea,0.139ml,0.8mmol)和4
‑
二甲氨基吡啶(dmap,2.5mg,0.02mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二异丙酯(33ul,0.2mmol)的ccl4(2ml)的溶液,缓慢升至20℃,薄层色谱(甲醇:二氯甲烷=1:5)监控反应。反应完全后减压浓缩,柱层析方法(硅胶柱提纯)纯化,既可以得到目标化合物5
‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二丙酯,其具体反应如式(9)所示:
[0085][0086]5‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二异丁酯为黄色固体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ12.28(s,1h),7.80(d,j=8.2hz,2h),6.95(t,j=17.8hz,2h),6.87(s,1h),5.44(s,1h),5.12(s,1h),4.77(dq,j=12.4,6.2hz,2h),4.48(s,1h),3.93(s,1h),3.81(s,3h),3.47(dd,j=28.8,14.3hz,3h),3.31(s,1h),1.68(d,j=22.0hz,6h),1.34(dd,j=18.9,6.1hz,12h),0.95(dd,j=12.2,6.1hz,3h);
31
p nmr(162mhz,cdcl3)δ
‑
8.96(s)。
[0087]
实施例10
[0088]5‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二异丁酯的结构式:
[0089][0090]
将实施例6合成的淫羊藿次苷ⅱ(103mg,0.2mmol)溶于乙醇(20ml),加入催化剂二异丙基乙胺(dipea,0.139ml,0.8mmol)和4
‑
二甲氨基吡啶(dmap,2.5mg,0.02mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二异丁酯(39ul,0.2mmol)的ccl4(2ml)的溶液,缓慢升至20℃,薄层色谱(甲醇:二氯甲烷=1:5)监控反应。反应完全后减压浓缩,柱层析方法(硅胶柱提纯)纯化,既可以得到目标化合物5
‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二异丁酯,其具体反应如式(10)所示:
[0091][0092]5‑
羟基
‑2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑7‑
磷酸二异丁酯为黄色固体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ12.39(s,1h),7.79(d,j=7.8hz,2h),6.96(d,j=7.8hz,2h),6.84(s,1h),5.43(s,1h),5.10(d,j=20.7hz,1h),4.47(s,1h),3.95(t,j=6.2hz,1h),3.90(dd,j=14.8,8.0hz,4h),3.81(s,3h),3.47(s,3h),3.31(s,1h),2.06
–
1.90(m,2h),1.68(d,j=21.7hz,6h),0.94(d,j=6.6hz,15h);
31
p nmr(162mhz,cdcl3)δ
‑
6.98(s)。
[0093]
实施例11
[0094]2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二甲酯的结构式为:
[0095][0096]
将实施例6合成的淫羊藿次苷ⅱ(103mg,0.2mmol)溶于乙醇(20ml),加入催化剂二异丙基乙胺(dipea,0.139ml,0.8mmol)和4
‑
二甲氨基吡啶(dmap,2.5mg,0.02mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二甲酯(36ul,0.4mmol)的ccl4(2ml)的溶液,缓慢升至30℃,薄层色谱(甲醇:二氯甲烷=1:5)监控反应。反应完全后减压浓缩,柱层析方法(硅胶柱提纯)纯化,得到目标化合物2
‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二甲酯,其具体反应如式(11)所示:
[0097][0098]2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二甲酯为黄色固体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ7.82(d,j=8.3hz,2h),7.36(s,1h),7.00(d,j=8.3hz,2h),5.48(d,j=10.0hz,1h),5.15(d,j=6.5hz,1h),4.45(s,1h),4.20
–
4.06(m,2h),4.00
–
3.84(m,16h),3.80
–
3.77(m,3h),3.57(d,j=20.7hz,2h),1.71(d,j=14.9hz,6h),1.40
–
1.30(m,3h);
31
p nmr(162mhz,cdcl3)δ
‑
4.65(s),
‑
5.05(s)。
[0099]
实施例12
[0100]2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二乙酯的结构式为:
[0101][0102]
将实施例6合成的淫羊藿次苷ⅱ(103mg,0.2mmol)溶于乙醇(20ml),加入催化剂二异丙基乙胺(dipea,0.139ml,0.8mmol)和4
‑
二甲氨基吡啶(dmap,2.5mg,0.02mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二乙酯(50ul,0.4mmol)的ccl4(2ml)的溶液,缓慢升至30℃,薄层色谱(甲醇:二氯甲烷=1:5)监控反应。反应完全后减压浓缩,柱层析方法(硅胶柱提纯)纯化,得到目标化合物2
‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二乙酯,其具体反应如式(12)所示:
[0103][0104]2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二乙酯为黄色固体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ7.83(d,j=8.6hz,2h),7.40(s,1h),7.00(d,j=8.6hz,2h),5.43(s,1h),5.15(t,j=6.2hz,1h),4.46(s,1h),4.35(dd,j=14.8,7.2hz,4h),4.24(ddd,j=15.1,7.3,3.5hz,4h),3.85(s,3h),3.59(s,2h),1.74
–
1.66(m,6h),1.37(t,j=6.9hz,12h),0.93(d,j=6.1hz,3h);
31
p nmr(162mhz,cdcl3)δ
‑
7.03(s),
‑
7.47(s)。
[0105]
实施例13
[0106]2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二异丙酯的结构式为:
[0107][0108]
将实施例6合成的淫羊藿次苷ⅱ(103mg,0.2mmol)溶于乙醇(20ml),加入催化剂二异丙基乙胺(dipea,0.139ml,0.8mmol)和4
‑
二甲氨基吡啶(dmap,2.5mg,0.02mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二异丙酯(66ul,0.4mmol)的ccl4(2ml)的溶液,缓慢升至30℃,薄层色谱(甲醇:二氯甲烷=1:5)监控反应。反应完全后减压浓缩,柱层析方法(硅胶柱提纯)纯化,得到目标化合物2
‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二异丙酯,其具体反应如式(13)所示:
[0109][0110]2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二异丙酯为黄色固体,其核磁共振光谱为:1h nmr(400mhz,dmso)δ7.87(d,j=8.3hz,2h),7.44(s,1h),7.13(d,j=8.2hz,2h),5.23(s,1h),5.15(s,1h),4.98(s,1h),4.88
–
4.80(m,2h),4.78(s,1h),4.70(dt,j=12.4,6.2hz,2h),4.00(s,1h),3.86(s,3h),3.65
–
3.43(m,5h),3.21
–
3.05(m,2h),1.67(d,j=24.0hz,6h),1.42
–
1.16(m,24h),0.80(d,j=4.6hz,3h);
31
p nmr(162mhz,cdcl3)δ
‑
8.92(s),
‑
8.96(s)。
[0111]
实施例14
[0112]2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二异丁酯的结构式为:
[0113][0114]
将实施例6合成的淫羊藿次苷ⅱ(103mg,0.2mmol)溶于乙醇(20ml),加入催化剂二异丙基乙胺(dipea,0.139ml,0.8mmol)和4
‑
二甲氨基吡啶(dmap,2.5mg,0.02mmol),冰水浴中降温至0℃,缓慢加入亚磷酸二异丁酯(80ul,0.4mmol)的ccl4(2ml)的溶液,缓慢升至30℃,薄层色谱(甲醇:二氯甲烷=1:5)监控反应。反应完全后,减压浓缩,柱层析方法(硅胶柱提纯)纯化,得到目标化合物2
‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二异丁酯,其具体反应如式(14)所示:
[0115][0116]2‑
(4
‑
甲氧基苯基)
‑8‑
(3
‑
甲基丁
‑2‑
烯
‑1‑
基)
‑3‑
(((3s,4s,5s,6r)
‑
3,4,5
‑
三羟基
‑6‑
甲基四氢
‑
2h
‑
吡喃
‑2‑
基)氧)
‑
4h
‑
色烯
‑4‑
酮
‑
5,7
‑
二磷酸二异丁酯为黄色液体,其核磁共振光谱为:1h nmr(400mhz,cdcl3)δ7.86
–
7.82(m,2h),7.38(s,1h),7.02(t,j=5.9hz,2h),5.44(d,j=1.3hz,1h),5.16(t,j=6.6hz,1h),4.45(s,1h),4.06(td,j=6.6,4.5hz,4h),3.97
–
3.90(m,5h),3.87(s,3h),3.56(d,j=25.0hz,3h),3.44(t,j=8.1hz,1h),3.33(dq,j=12.3,6.1hz,1h),2.01
–
1.95(m,4h),1.73(s,3h),1.69(s,3h),1.26(s,3h),0.96(dd,j=6.5,4.6hz,24h);
31
p nmr(162mhz,cdcl3)δ
‑
6.84(s),
‑
7.35(s)。
[0117]
实施例15淫羊藿素和淫羊藿次苷ⅱ衍生物对raw264.7(小鼠单核巨噬细胞白血病细胞)的影响
[0118]
1.将raw264.7(小鼠单核巨噬细胞白血病细胞)进行细胞复苏1000r/min离心5min弃上清。完全培养基重悬,吹打均匀,接种到培养瓶中培养,放入37℃,5%co2培养箱中恒温培养,每三天观察并换液一次。待细胞长满后,进行传代或者种板。
[0119]
2.完全培养基:rpmi1640,10%胎牛血清,1%双抗培养瓶中的细胞生长到80%~90%时,进行细胞毒实验,用0.25%胰蛋白酶溶液将细胞消化后,离心除去胰蛋白酶,使用3ml的完全培养基进行重悬,吹打均匀,调整细胞密度为5x104个/ml接种到96孔板中,每孔
100ul,培养24h后。以dmso为阴性对照组,药物作用48h后,进行细胞活力实验,实验结果如表1所示:
[0120]
3.细胞活力实验:使用mtt比色法检测细胞存活,其检测原理是活细胞线粒体中的琥珀酸脱氢酶能使外源性mtt还原为水不溶性的蓝紫色结晶甲瓒并沉积在细胞中,而死细胞无此功能。二甲基亚砜(dmso)能溶解细胞中的甲瓒,用酶标仪测量490nm波长的吸光度值,可间接反映活细胞数量。
[0121]
表1淫羊藿素和淫羊藿次苷ⅱ衍生物对raw264.7(小鼠单核巨噬细胞白血病细胞)的细胞存活率
[0122][0123][0124]
表1为淫羊藿素和淫羊藿次苷ⅱ衍生物对raw264.7(小鼠单核巨噬细胞白血病细胞)的细胞存活率。从表1可知本发明的淫羊藿次苷ⅱ衍生物,在raw264.7细胞毒实验中显示,淫羊藿次苷ⅱ磷酸二甲酯的细胞毒性较淫羊藿素和淫羊藿次苷ⅱ显著降低,说明磷酸甲酯的修饰有利于降低对正常细胞的毒性。
[0125]
实施例16淫羊藿素和淫羊藿次苷ⅱ衍生物的对pc
‑
3细胞的细胞毒实验
[0126]
1.将pc
‑
3(人前列腺癌细胞)进行细胞复苏,1000r/min离心5min弃上清。完全培养基重悬,吹打均匀,接种到培养瓶中培养,放入37℃,5%co2培养箱中恒温培养,每三天观察并换液一次。待细胞长满后,进行传代或者种板。
[0127]
2.完全培养基:rpmi1640,10%胎牛血清,1%双抗培养瓶中的细胞生长到80%~90%时,进行细胞毒实验,用0.25%胰蛋白酶溶液将细胞消化后,离心除去胰蛋白酶,使用3ml的完全培养基进行重悬,吹打均匀,调整细胞密度为5x104个/ml接种到96孔板中,每孔100ul,培养24h后。以dmso为阴性对照组,药物作用24h后,进行细胞活力实验。实验结果如表3所示:
[0128]
表2淫羊藿素和淫羊藿次苷ⅱ磷酸酯化衍生物对pc
‑
3的半抑制率
[0129][0130]
注:空白对照组比较,p值均小于0.05;ic50:即半抑制浓度,或称半抑制率
[0131]
表2为淫羊藿素和淫羊藿次苷ⅱ磷酸酯化衍生物的对pc
‑
3的半抑制率。从表2可知,淫羊藿次苷ⅱ衍生物在pc
‑
3细胞毒实验中显示,淫羊藿次苷ⅱ衍生物(除磷酸二甲酯衍生物外)的细胞毒性显著增强,在同等浓度下,可以用于制备预防或治疗癌症的药物。
[0132]
实施例17淫羊藿素的磷酸酯化衍生物对成骨细胞矿化结节形成的影响
[0133]
1.成骨细胞培养:将ec3t3
‑
e1 subclone 14(小鼠颅顶前骨细胞亚克隆14)进行细胞复苏,1000r/min离心5min弃上清。完全培养基(完全培养基:α
‑
mem,10%胎牛血清,1%双抗)重悬,吹打均匀,接种到培养瓶中培养放入37℃,5%co2培养箱中恒温培养,每三天观察并换液一次。待细胞长满后,进行传代或者种板。
[0134]
2.成骨细胞矿化结节形成测定:待培养瓶中的细胞生长到80~90%融合时进行细胞矿化结节形成实验。用胰酶将细胞消化后,离心除去胰酶,使用3ml的完全培养基进行重悬,吹打均匀,调整细胞密度为5x104个/ml接种到24孔板中,每孔500ul。培养24h细胞贴壁后,分别换完全培养基、成骨分化培养基和含药物的成骨诱导培养基(成骨诱导培养基:α
‑
mem,10%胎牛血清,1%双抗,10mmolβ
‑
甘油磷酸钠,10nmol地塞米松,50ug/ml抗坏血酸细胞)继续培养。培养18天后进行矿化结节实验:先除去培养基,然后细胞用pbs洗3次,每次5min,每孔加入500ul的4%多聚甲醛对细胞进行固定,室温固定细胞30min,然后pbs洗3次,每次5min,每孔加入500ul的茜素红染液,茜素红能够与钙离子螯合,生成橘红色沉淀,能够检测细胞外钙离子的形成和定量研究。37℃染色30min,然后用pbs清洗至清洗液不含颜色,显微镜拍照,实验结果如图1所示,图1为实施例1
‑
4中淫羊藿素的磷酸酯化衍生物对成骨细胞矿化结节的影响。其中,a为完全培养基(不含药物)的茜素红染色的显微照片;b为空白成骨培养基(不含药物)的茜素红染色的显微照片;c
‑
j:分别是实施例1(62.5nmol/l、实施例1(500nmol/l)、实施例2(62.5nmol/l)、实施例2(500nmol/l)、实施例3(62.5nmol/l)、实施例3(500nmol/l)、实施例4(62.5nmol/l)和实施例4(500nmol/l)的茜素红染色的显微拍照。从图1中可知,本发明的实施例1
‑
4中在淫羊藿素的磷酸酯化衍生物在较低浓度(62.5~500nmol/l)浓度下能明显促进成骨细胞形成钙结节的能力。
[0135]
本发明淫羊藿素的磷酸酯化衍生物在62.5~500nmol/l浓度下能明显促进成骨细
胞形成钙结节的能力,说明该淫羊藿素的磷酸酯化衍生物具有促进成骨细胞矿化的能力,促进成骨形成的优点,用于预防和治疗骨质疏松症具有一定的应用前景。
[0136]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合和简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1