一种蛋白降解靶向嵌合体的制备方法和应用
1.本发明涉及医药技术领域,具体为一种蛋白降解靶向嵌合体的制备方法和应用,以及该类小分子化合物在治疗恶性肿瘤疾病中的应用。
背景技术:
2.protac作为一种新兴的技术,通过双功能小分子使靶蛋白被e3泛素连接酶识别并打上泛素化标签,然后利用细胞自身的泛素-蛋白酶体系统降解靶蛋白。目前,大多数有关protac的研究都集中在降解单个靶标蛋白上。尽管有一些文献中已经发表了可以实现两种或多种蛋白的下调,但大多数是由protac脱靶效应引起的,或者是由于抑制了上游信号引起下游多种蛋白下调的结果,或由于作用于相同类型蛋白的配体口袋导致,因此protac分子可以同时实现下调多个相同类型的靶蛋白,例如目前报道对激酶的降解。目前,尚未实现使用protac技术将不同类型的靶蛋白进行同步双重敲除。这方面的主要挑战是不同类型蛋白质的配体结合口袋非常不同,并且设计可同时结合不同类型靶蛋白的配体更加困难。其次,因为protac化合物是一种双功能分子介导的三元复合物,复合物形成和识别的动态过程涉及e3连接酶与靶蛋白之间的相互作用,因此对双功能分子本身的大小和结构都有严格的要求。利用小分子选择性地使不同类型的靶蛋白进行同步协同敲除,不仅将产生具有发展潜力的治疗方法,而且还将提供有价值的工具,进行化学和生物学相关的研究。
3.bet家族在人体组织广泛表达,与多种疾病的发生和发展密切相关,近年来研究发现,bet蛋白参与睾丸核蛋白中线癌(nut midline carcinoma,nmc)、急性白血病、淋巴瘤、乳腺癌等肿瘤的发生、浸润和转移过程。bet家族成员brd4可调节癌基因myc,bcl2和bcl6等转录过程。抑制或敲除brd4,可诱发肿瘤细胞凋亡,从而达到抗肿瘤作用;组蛋白去乙酰化酶(hdacs)是一种重要的表观遗传学清除剂,可从组蛋白的赖氨酸上除去乙酰基。它们包含11个组分,分为4类(i,iia,iib和iv)和7个沉默调节蛋白(iii类)。在癌细胞中,hdacs的过度表达导致去乙酰化作用增强,不利于特定基因的表达,包括一些肿瘤抑制基因。迄今为止,已经批准了五种hdac抑制剂 (hdacis),即伏立诺他(saha),罗米地辛(fk228),贝利诺司他(pxd-101),帕诺比司他(lbh-589)和甲胺酰胺,用于治疗血液癌和胰腺癌。brd4与hdacs 这两个靶标在部分肿瘤的发生和发展中都起着重要作用,均是癌症和其他慢性疾病中的表观遗传学靶标,已有研究报道同时抑制brd4和hdacs可以协同下调c-myc等癌基因的表达,达到协同抗肿瘤的效果。本课题组前期通过药效团融合策略设计合成了一系列新颖的bet/hdacs双靶抑制剂,在体外与体内都展现了出色的抗肿瘤效果。但这类双靶抑制剂仍具有一般抑制剂的某些局限性,例如严重的脱靶毒性和更高剂量的依赖性。从理论上说,使用 protac实现brd4和hdacs的双重敲除将克服双靶抑制剂所存在的问题,目前尚无文献报道使用合理设计的protac同时对两种靶蛋白的双重敲除。基于上述原因,我们使用protac技术设计出了能够将brd4和hdacs进行同步敲除的双靶protac分子。
技术实现要素:
4.本发明的目的是提供一类基于brd4蛋白与hdacs蛋白降解剂及其药学上可接受的盐。本发明还提供该化合物的制备方法和应用。
5.本发明的技术方案如下:
6.1、通式(i)的化合物及药学上可接受的盐:
[0007][0008]
x为1至60个碳原子的饱和或不饱和的直链烃基、氧杂链、间氧杂链、苯基、杂环基团或以下链接基团之一:
[0009][0010]
y为1至60个碳原子的饱和或不饱和的直链烃基、氧杂链、间氧杂链、苯基、杂环基团或以下链接基团之一:
[0011]
[0012][0013]
其中n是0-30。
[0014]
其中,优选的杂环基团为哌嗪基、吡咯基、吡唑基、呋喃基、噻吩基、噁唑基、异噁唑基、噻唑基、异噻唑基、吡啶基、嘧啶基、吡嗪基或哒嗪基或以下链接基团之一:
[0015][0016]
其中n是0-30。
[0017]
其中,优选的杂环基团为哌嗪酮基、吡咯基、吡唑基、呋喃基、噻吩基、噁唑基、异噁唑基、噻唑基、异噻唑基、吡啶基、嘧啶基、吡嗪基或哒嗪基。 a或b为卤素、烯烃、醇、酚、醚、醛、酮、羧酸、硝基、磺酸类有机物、胺类、酰胺类、酯基或下列基团:-x、-oh、-cho、-cooh、-no2、-so3h、-nh2、-co-[0018][0019]
其中n是0-30。
[0020]
所述的药学上可接受的盐包括通式i化合物与下列酸形成的酸加成盐:盐酸、氢溴酸、硫酸、乳酸、柠檬酸、磷酸、甲磺酸、苯磺酸、对甲苯磺酸、萘磺酸、酒石酸、丙酮酸、乙酸、马来酸或琥珀酸、富马酸、水杨酸、苯基乙酸、杏仁酸。
[0021]
根据本发明,进一步优选的,上述通式(i)化合物是下列之一:
[0022]
8-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2-f][1,2,4] 三唑[4,3-a][1,4]二氮杂-1-基)-n-(2-(2-(2-(2-((2-(2,6-二氧代哌啶-3-基)1-氧代异吲哚-4-基)氨基)-2-氧乙氧基)乙氧基)乙氧基)乙基)乙酰胺基)-n-羟基辛酰胺(m10)
[0023]
8-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2-f][1,2,4] 三唑[4,3-a][1,4]二氮杂-6-6基)-n-(2-(2-(2-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚-4-基]
氨基)-2-氧代乙氧基)乙氧基)乙基) 乙酰氨基)辛酸(m12)
[0024]
8-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2-f][1,2,4] 三唑[4,3-a][1,4]二氮杂-1-基)-n-(2-(3-((2-(2,6-二氧代哌啶
‑ꢀ
3-基)-1-氧代异吲哚-4-基)氨基)-3-氧代丙氧基)乙基)乙酰胺基)辛酸(m14)
[0025]
14-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2-f][1,2,4] 三唑[4,3-a][1,4]二氮杂-6-6基)-n-(8-(羟基氨基)-8-氧辛基)乙 酰氨基)-n-(2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-4-基)
ꢀ‑
3,6,9,12-四氧杂环丁酸酯(m16)
[0026]
17-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2-f][1,2,4] 三唑[4,3-a][1,4]二氮杂-6-6基)-n-(8-(羟基氨基)-8-氧辛基)乙酰氨基)-n-(2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-4-基)
ꢀ‑
3,6,9,12,15-五氧杂十八碳酰胺(m18)
[0027]
20-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2-f][1,2,4] 三唑[4,3-a][1,4]二氮杂-6-6基)-n-(8-(羟基氨基)-8-氧辛基)乙酰氨基)-n-(2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-4-基)
ꢀ‑
3,6,9,12,15,18-六氧杂二十二酰胺(m20)
[0028]
以上优选化合物,后面的括号内的编号是对应于下面反应路线及表中的化合物结构的编号。
[0029]
本发明的化合物制备方法如下:
[0030]
通法:
[0031][0032]
通法:
[0033]
5,8,11-三氧杂-2-氮杂十三烷二酸-1-叔丁酯(m0)与来那度胺(m1) 在dmf中进行
缩合反应得到化合物m2,
[0034]
m2在三氟乙酸存在的条件下脱去boc保护基得到化合物m3,m3与m4进行取代反应得到化合物m5,
[0035]
m5与(+)-jq1羧酸进行缩合反应得到化合物m6,
[0036]
m6在碱性条件下进行酯交换反应得到m7,
[0037]
m7在三氟乙酸条件下脱去羧基保护基叔丁酯得到化合物m8,
[0038]
m8与o-三苯甲基羟胺进行缩合反应得到化合物m11,
[0039]
m11最后在三氟乙酸存在的条件下脱去保护基暴露出异羟肟酸基团得到化合物m10。
[0040]
其中hatu为2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯, dipea为n,n-二异丙基乙胺,meoh为甲醇,ch2cl2为二氯甲烷,dmf为二甲基甲酰胺,tea为三乙胺以下含义类同。
[0041]
化合物m1与其他原料通过市场购买;
[0042]
本发明也提供了上述结构通式(i)所示化合物在制备brd4降抑制剂或hdacs抑制剂中的应用。
[0043]
本发明还提供了上述结构通式(i)所示化合物在治疗与上述靶点有关的肿瘤疾病中的应用,包括对肿瘤细胞的杀伤作用。与brd4或hdacs有关的疾病可以是,但不限于:肺癌、肝癌、肾癌、非小细胞肺癌、前列腺癌、甲状腺癌、皮肤癌、胰腺癌、卵巢癌、乳腺癌、膀胱癌、骨髓增生异常综合症、淋巴瘤、食管癌、胃肠道癌、骨肉瘤、中枢或外周神经系统的肿瘤。
附图说明
[0044]
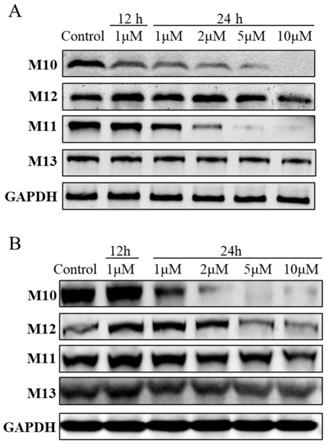
图1为本发明提出的一种蛋白降解靶向嵌合体的制备方法和应用的分析结果示意图。
具体实施方式
[0045]
现结合实施例,对本发明作详细描述,但本发明的实施不仅限于此。本发明所用试剂和原料均市售可得或可按文献方法制备。
[0046]
下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。
[0047]
以下实施例所涉化合物的1hnmr,
13
cnmr和ms数据详见表1.1。
[0048]
表1.1优选化合物1hnmr,
13
cnmr和ms数据
[0049]
[0050]
[0051]
[0052]
[0053][0054]
实施例1:
[0055]
8-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2-f] [1,2,4]三唑[4,3-a][1,4]二氮杂-1-基)-n-(2-(2-(2-(2-((2
‑ꢀ
(2,6-二氧代哌啶-3-基)1-氧代异吲哚-4-基)氨基)-2-氧乙氧基)乙氧基)乙氧基)乙基)乙酰胺基)-n-羟基辛酰胺的合成(m10)
[0056]
步骤a:叔丁基(2-(2-(2-(2-(2-((2-(2,6-二氧代哌啶-3-基)1
‑ꢀ
氧代异吲哚基-4-基)氨基)-2-氧代乙氧基)乙氧基)乙氧基)氨基甲酸乙酯的合成(m2)
[0057]
步骤a:将化合物m0(1.0g,3.1mmol)h,atu(2.4g,6.2mmol)d,ipea(1.0 ml,7.8mmol)溶于干燥的dmf中,在室温条件下反应0.5h然后加入来那度胺m1(0.80g,3.1mmol)后反应4h,经tlc检测反应完全。将反应液缓慢倒入冰水(1.5l)混合物中,用ea(30ml
×
3)萃取后合并有机相,减压蒸除溶剂,剩余物用硅胶柱色谱分离,流动相为dcm/meoh=100/5,得
到黄色油状液体m2(1.2g,2.2mmol,产率:70%)。
[0058]
步骤b:2-(2-(2-(2-(2-氨基乙氧基)乙氧基)乙氧基)-n-(2
‑ꢀ
(2,6-二氧代哌啶-3-基)-1-氧代异吲哚-4-基)乙酰胺的合成(m3)
[0059]
步骤b:将化合物m2(600mg,1.1mmol)溶于干燥的二氯甲烷(8.0ml) 中加入tfa(2.0ml)在室温条件下反应2h后经tlc检测反应较完全,减压蒸除溶剂,剩余物无需处理直接投下一步反应。
[0060]
步骤c:1-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚-4-基)氨基)-1-氧代-3,6,9-三氧杂-12-氮杂二十烷酸乙酯的合成(m5)
[0061]
将化合物m4(203mg,0.81mmol),k2co3(224mg,1.6mmol),ki(6.7 mg,0.041mmol)溶于干燥的乙腈中,在40℃条件下反应十分钟后加入化合物m3(363mg,0.81mmol)反应12h,经tlc检测反应完全。将溶剂减压蒸除,剩余物用硅胶柱色谱分离,流动相为dcm/meoh=100/7,得到无色透明液体m5(150mg,0.24mmol,产率:30%)。
[0062]
步骤d:乙基12-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂卓-6-基)乙酰基)-1
‑ꢀ
((2-(2,6-二氧哌啶-3-基)-1-氧代异吲哚-4-基)氨基)-1-氧代-3,6,9
‑ꢀ
三氧代-12-氮杂-20-酸盐的合成(m6)
[0063]
步骤d:将化合物(+)-jq1羧酸(200mg,0.50mmol),hatu(380mg, 1.0mmol),dipea(162mg,1.3mmol)加入到干燥的dmf中,在室温条件下反应0.5h后加入化合物m4(309mg,0.5mmol)反应4h,经tlc检测反应完全。将反应液缓慢倒入冰水(500ml)混合物中,用ea(20ml
×
3)萃取后合并有机相,减压蒸除溶剂,剩余物用硅胶柱色谱分离流动相为 dcm/meoh=100/3,得到白色固体m6(500mg,0.40mmol,产率:80%)。
[0064]
步骤e:叔丁基12-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h
‑ꢀ
噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基)乙酰基)-1
‑ꢀ
((2-(2,6-二氧哌啶-3-基)-1-氧代异吲哚-4-基)氨基)-1-氧代-3,6,9
‑ꢀ
三氧代-12-氮杂-20-酸盐的合成(m7)
[0065]
步骤e:将化合物m5(100mg,0.080mmol),tbaf(21mg,0.080mmol) lioh(7.6mg,0.32mmol),溶于干燥的四氢呋喃中,在80℃条件下反应2 h后,加入化合物叔丁醇(30mg,0.40mmol)反应4h,经tlc检测反应较完全,减压蒸除溶剂,剩余物用硅胶柱色谱分离,流动相为dcm/meoh= 100/5,得到白色固体m7(58mg,0.056mmol,产率:70%)。
[0066]
步骤f:12-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2
‑ꢀ
f][1,2,4]三唑并[4,3-a][1,4]二氮杂卓-6-基)乙酰基)-1-((2-(2,6
‑ꢀ
二氧哌啶-3-基)-1-氧代异吲哚-4-基)氨基)-1-氧代-3,6,9-三氧代-12
‑ꢀ
氮杂-20-甲酸的合成(m8)
[0067]
步骤f:将化合物m7(100mg,0.097mmol)溶于干燥的二氯甲烷(8.0 ml)中,加入tfa(2.0ml)在室温条件下反应4h,经tlc检测反应较完全,减压蒸除溶剂,剩余物无需处理直接投下一步反应。
[0068]
步骤g:8-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2
‑ꢀ
f][1,2,4]三唑[4,3-a][1,4]二氮杂-1-基)-n-(2-(2-(2-(2-((2
‑ꢀ
(2,6-二氧代哌啶-3-基)1-氧代异吲哚-4-基)氨基)-2-氧代乙氧基)乙氧基)乙氧基)乙基)乙酰胺基)-n-(三苯甲氧基)辛酰胺的合成(m11)
[0069]
步骤g:将化合物m8(50mg,0.051mmol),hatu(39mg,0.102mmol), dipea(16mg,0.13mmol)溶于干燥的dmf中,在室温条件下反应0.5h后,加入化合物o-三苯甲基羟胺
(72mg,0.26mmol)反应2h,经tlc检测反应完全,将反应液缓慢倒入冰水(30ml)混合物中用ea(10ml
×
3)萃取后合并有机相,减压蒸除溶剂,剩余物用硅胶柱色谱分离,流动相为dcm/meoh =100/5,得到白色固体m11(53mg,0.043mmol,产率:85%)。
[0070]
步骤h:8-(2-((s)-4-(4-氯苯基)-2,3,9-三甲基-6h-噻吩并[3,2
‑ꢀ
f][1,2,4]三唑[4,3-a][1,4]二氮杂-1-基)-n-(2-(2-(2-(2-((2
‑ꢀ
(2,6-二氧代哌啶-3-基)1-氧代异吲哚-4-基)氨基)-2-氧乙氧基)乙氧基)乙氧基)乙基)乙酰胺基)-n-羟基辛酰胺的合成(m10)
[0071]
步骤h:将化合物m11(53mg,0.043mmol)溶于干燥的二氯甲烷(8.0 ml)中,然后加入tfa(2.0ml)在室温条件下反应3h,经tlc检测反应完全,减压蒸除溶剂,剩余物用硅胶柱色谱分离,流动相为dcm/meoh= 100/10,得到白色固体m10(39mg,0.039mmol,产率:90%)。
[0072]
化合物m12,m14,m16,m18,m20的合成过程参照化合物m10的合成步骤。
[0073]
实施例2:本发明化合物对brd4与hdac1抑制活性测试(ki)。
[0074]
将5μl待测化合物(各稀释浓度)、brd4(20nm)及pmdm6-f(20nm) (缓冲液:100mm磷酸三钾,ph=7.5;100μg/ml bgg;0.02%叠氮钠)加入96孔黑色平底酶标板至终体积115μl,室温孵育1小时后,用biotek
‑ꢀ
synergy酶标仪(激发光为485nm,发射光为528nm)读数荧光偏振值。
[0075]
依据上述方法得到的荧光偏振值,用origin 9.0软件绘制曲线,计算得蛋白结合抑制常数(ki)。hdac1测试方法同brd4。
[0076]
实验结果:首先测试所有目标化合物对brd4和hdac1蛋白的抑制活性,选用(+)-jq1与saha作为阳性对照。测试结果如表1.2 所示,保留与brd4和hdacs蛋白结合活性基团的化合物m10对两种蛋白均有较强的抑制活性;对与brd4产生结合作用的基团进行封闭只保留与hdacs产生结合作用的基团,化合物m12展现出对 hdac1的较强抑制活性,对brd4没有抑制活性;对与hdacs产生结合作用的基团进行封闭只保留与brd4产生结合作用的基团,化合物m11展现出对brd4的较强抑制活性,对hdac1没有抑制活性;对与brd4和hdacs都产生结合作用的基团进行封闭的化合物 m13对两种蛋白都没有抑制活性。通过封闭能够与靶蛋白产生结合的活性基团,初步验证了bet/hdac双靶protac化合物m10对两种靶蛋白都能产生抑制活性,说明m10能够与靶蛋白上的活性位点结合,这是protac分子发挥降解功能的基础与预期结果相符合。
[0077]
表1.2目标化合物对brd4和hdac1的抑制活性(ic
50
,μm)
[0078][0079]
实施例3:本发明化合物的体外抗肿瘤活性测试(ic
50
)。
[0080]
对本发明的化合物进行了三种肿瘤细胞增殖抑制能力测试,测试方法采用常规的cck8法。将对于对数生长期的肿瘤细胞(mcf-7 (人乳腺癌细胞))、a549(人肺癌细胞)、
hepg2(人肝癌细胞)用胰酶消化,然后用培养基(dmem+10%fbs或者prmi1640+10%fbs) 将细胞稀释悬浮成单细胞悬液,调整细胞密度为5
×
104个/ml,每孔加入100μl接种于96孔板中,置37℃、5%co2培养箱内培养24 小时,再分别加入不同浓度的化合物,每个浓度平行三个复孔,并设置实验组和对照组,继续孵育72小时后,向每孔加入10μl cck8 溶液,然后37℃下避光孵育1-4小时后,用biotek-synergy酶标仪测 450nm od值。计算半数抑制浓度ic
50
。
[0081]
实验结果:进一步评价目标化合物的体外抗肿瘤活性,选用a549(人肺癌细胞)、hepg2(人肝癌细胞)、mcf-7(人乳腺癌细胞)作为测试肿瘤株,(+)-jq1与saha作为阳性对照。测试结果如表1.3所示,整体看化合物m10在所有目标化合物中抗肿瘤活性最好比阳性药稍好;m13在所有化合物中抗肿瘤活性最差;m11与m12抗肿瘤活性介于m10与m13之间,目标化合物的体外抗肿瘤活性与化合物对 brd4和hdac1抑酶活性基本保持一致,抑酶活性与细胞活性结果均显示,化合物m10在所有目标化合物中表现出最好的活性,达到化合物的设计要求,下面对目标化合物开展作用机制研究,进一步验证bet/hdac双靶protac的设计思想。
[0082]
表1.3.目标化合物的体外抗肿瘤活性(ic50,μm)a
[0083]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1