一种分离纯化红景天苷的方法及其产物与流程
1.本发明涉及有机物分离提取技术领域,具体地说,涉及一种分离纯化红景天苷的方法及其产物。
背景技术:
2.红景天为多年生草本植物,主要生长在海拔1600~4000米的高寒、干燥、缺氧、强紫外线照射、昼夜温差大的地区,具有极强的环境适应能力和生命力。在《四部医典》和《本草纲目》均有记载,研究证明红景天苷有抗疲劳、抗衰老、免疫调节、清除自由基等多种药理作用。
3.在安普胶囊的成分中,红景天苷也有相应作用。红景天苷能够干扰细胞代谢、改变细胞外衣的性质,抑制肿瘤细胞增殖的同时,还能够提高t淋巴细胞转化率和吞噬细胞活力,增强免疫力,抑制肿瘤生长,使白血球升高,抵抗微波辐射等作用。
4.因此,红景天苷(salidroside)作为药物或功能食品具有很好的应用前景。其结构式如下所示:
[0005][0006]
目前,红景天苷的生产方法主要是化学合成法及发酵法,化学合成法步骤多、副产物多且难以去除。
[0007]
现阶段重点则为从红景天中提纯红景天苷,已经报道的有使用葡聚凝胶吸附解吸,模拟移动床层析,逆流萃取,大孔树脂吸附,醇解吸,反相c18制备色谱分离,复合酶解,混合溶剂结晶等提取方法。
[0008]
中国专利cn103467540a中采用大孔树脂吸附,高浓度醇洗脱,洗脱液浓缩,无水乙醇溶解再经过模拟移动床色谱分离,结晶。过程中经过1次层析和模拟移动床色谱分离,周期长,过程使用有机溶剂,成本和安全性需要考虑。
[0009]
cn105085588a使用硅胶柱色谱富集,微孔树脂除杂,反相液相制备色谱精制,全程使用有机溶剂成本高。
[0010]
cn106117281a,cn108727441a,cn109929002a,cn107686492a,cn101392011等专利均采用大孔树脂吸附,有机溶剂解吸。cn105924481a采用生物酶法,存在酶的失活及回收问题,同时连续逆流提取也使用大量有机溶剂,cn106279310a采用乙醚,cn104892696a采用丙酮,安全性难以保障。美国专利us201816224257公布了转基因菌种生产红景天苷的原理及基因构建方法和分离纯化方法,70%乙醇提取,浓缩,分别正己烷、氯仿、丁醇依次提取,lh20分离,水
‑
甲醇梯度为0
‑
100%纯化。目前公开的红景天苷提取方法几乎都采用大孔树脂有机溶剂层析和色谱分离或者制备色谱有机溶剂色谱分离,存在成本问题和安全性风险,故有必要提供一种新的分离纯化红景天苷的方法。
技术实现要素:
[0011]
针对现有技术的问题,本发明的目的是提供一种可快速简便、成本低廉(不使用有机溶剂)的制备高纯度红景天苷的工艺技术。
[0012]
为了实现该目的,本发明的技术方案如下:
[0013]
一种分离纯化红景天苷的方法,所述方法不采用有机溶剂;包括将红景天苷发酵液在ph 3~5下经大孔树脂吸附后,以碱性缓冲盐溶液进行洗脱的步骤。
[0014]
本发明所述红景天苷发酵液为以微生物发酵形式产生的含有红景天苷的发酵液。优选在大孔树脂吸附后先进行水洗,再以碱性溶液洗脱。
[0015]
本发明先在特定酸性条件下将红景天苷吸附在树脂上,并将极性较强物质以及色素、酸性沉淀的杂质留在清液中。水洗干净树脂后,再在特定碱性条件下进行解吸,从而将极性较弱的杂质和部分色素留在树脂上,实现有效的提纯,使产品纯度大幅提高,颜色去除较多。
[0016]
本发明中,所述碱性缓冲盐溶液的ph值为8~12,优选为9~11;
[0017]
所述碱性缓冲盐溶液中的碱性缓冲盐为碳酸氢铵,碳酸氢钠,磷酸铵,磷酸钠,枸橼酸钠或季铵盐,优选为磷酸钠盐,如磷酸二氢钠。其在洗脱液的ph条件下缓冲能力很强,ph值受物料波动影响小。
[0018]
本发明中,所述碱性缓冲盐溶液的浓度为0.25~1%;
[0019]
优选,所述洗脱为以0.5%的碱性缓冲盐溶液和0.75%的碱性缓冲盐溶液进行梯度洗脱,可实现理想的收率。
[0020]
本发明中,在洗脱后,还包括以活性炭在ph 2~5,40~80℃下进行处理的步骤。
[0021]
优选以活性炭在ph 2~4,50~60℃下进行处理。
[0022]
所述活性炭的加入量为0.1~0.5%(w/v),优选为0.3~0.5%(w/v)。
[0023]
本发明配合上述特定吸附和解吸步骤处理后的溶液的特点,设置了采用活性炭在特定酸性条件下进一步去除大分子物质及色素的步骤。该步骤可在高温条件下进行,能有效提升处理效果。
[0024]
本发明中,在活性炭处理后,还包括降温结晶的步骤;所述降温结晶步骤具体为:
[0025]
开始温度为60℃,以每小时降温1℃的速率降至40℃后,再以每20分钟降温1℃的速率降至5℃。
[0026]
以本发明特定直接水相降温结晶方式,既可保证产物的有效析出,又可提升产物纯度。
[0027]
本发明中,所述红景天苷发酵液在以大孔树脂吸附前的浓度为40~60mg/ml;在活性炭处理前的浓度为60~100mg/ml,优选60~70mg/ml;在降温结晶前的浓度为400mg/ml~700mg/ml,优选400~600mg/ml。
[0028]
浓度的调整可采用旋转蒸发方式。
[0029]
本发明中,所述红景天苷发酵液在以大孔树脂吸附前依次经过过滤、超滤和纳滤;所述过滤的孔径为50nm;所述超滤在ph 7~9下进行,采用的超滤膜为分子量2000da的聚醚砜膜;所述纳滤采用的纳滤膜为分子量100da的聚醚砜膜。
[0030]
过滤可采用陶瓷膜进行。
[0031]
本发明中,所述大孔树脂为日本三菱hp20树脂。
[0032]
优选地,本发明综合采用红景天苷的发酵液陶瓷膜过滤,滤液经过超滤纳滤后,过大孔吸附树脂吸附解吸,活性炭去杂质,浓缩,降温结晶干燥技术进行。
[0033]
本发明方法在降温结晶后还包括分离、减压干燥的步骤。
[0034]
本发明还提供一种红景天苷产品,其根据上述的方法制备得到。该产品为白色粉末,纯度>99%,含量>99%。
[0035]
本发明的有益效果至少在于:
[0036]
本发明提供了一种分离纯化红景天苷的方法,该方法不用色谱分离,只是单纯吸附解吸,周期短,全程不使用有机溶剂,节约成本,工艺操作简便。同时本发明方法的质量水平和收率水平均很高。可获得色谱纯度和含量均大于99%的红景天苷固体,适合工业化生产红景天苷。
附图说明
[0037]
图1为本发明实施例中所采用的红景天苷发酵液色谱图。
[0038]
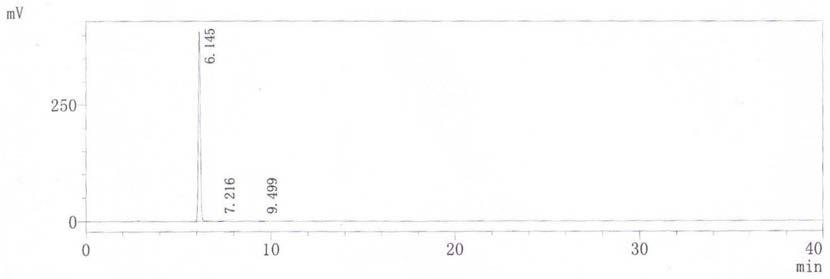
图2为本发明实施例2的终产品色谱图。
[0039]
图3为本发明实施例2的终产品外观图。
[0040]
图4为本发明对比例2中不同ph下活性炭处理后的滤液稀释至相同浓度后的照片,左侧试管为在ph 6.0下处理后的结果,中间试管为在ph5.0下处理后的结果,右侧试管为在ph 3.5下处理后的结果。
[0041]
图5为本发明对比例2中以活性炭在ph3.5下处理前后的浓缩液照片(浓度均为500mg/ml),左侧为处理前的,右侧为处理后的。
具体实施方式
[0042]
下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。
[0043]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0044]
本发明所用的富含红景天苷的原料来源于,一种转基因构建的菌种所发酵生产的发酵液。该转基因菌种生产红景天苷的原理及基因构建、发酵方法参见美国专利us201816224257(us 2019/0264221a1)(如第131段)。将该发酵液通过50nm陶瓷膜过滤得到富含红景天苷的陶瓷膜滤液。下面实施例、对比试验均采用此陶瓷膜过滤液。其色谱图参见图1,具体各峰检测信息见表1。色谱条件为:流动相∶甲醇
‑
水(15∶85);波长:223nm;溶剂:甲醇;柱:sdb c
‑
18;流速:1ml/min;浓度:1mg/ml。以下关于纯度测试时所采用的色谱条件与此相同。
[0045]
表1
[0046]
peak#ret.timeareaheightarea%13.82752505560.35325.127117554912130079.00835.8681499414801.008
46.2345933853203.98857.8891562881132010.50468.91049342180.33279.8033934622522.644811.95485013780.571913.34156732410.3811019.26649781680.3351135.995130282110.876总计 1487881143445100.000
[0047]
实施例1
[0048]
本实施例提供一种本发明的红景天苷分离提取方法,具体如下:
[0049]
将红景天苷陶瓷膜滤液调节至ph8.0,以分子量2000da的聚醚砜膜进行超滤,以分子量100da的聚醚砜膜进行纳滤后收浓为浓度50mg/ml的浓缩液,将浓缩液调节至ph4,加入hp20树脂静态搅拌室温条件下吸附2h,过筛,水洗干净后树脂装柱,用0.5%磷酸氢二钠
‑
ph10.5溶液和0.75%磷酸氢二钠
‑
ph10.5溶液(各5倍体积)进行梯度洗脱,收集洗脱液,经色谱测试,洗脱液纯度为:93.72%,收率为92.10%。将洗脱液旋转蒸发浓缩至浓度为60mg/ml,调节至ph3.5,60℃下加入活性炭0.3%(质量体积比)搅拌除杂30分钟,过滤,滤液继续旋转蒸发浓缩至浓度为500mg/ml,取出,60℃开始按照每小时降温1℃至40℃后,每20分钟降温1℃速率降温至5℃,分离,得到白色晶体,减压干燥(
‑
0.09mpa,60℃,20h),成品经检测,色谱纯度为:99.55%,含量:99.22%,收率为94.07%。最终整体收率为80.44%。
[0050]
实施例2
[0051]
本实施例提供一种本发明的红景天苷分离提取方法,具体如下:
[0052]
将红景天苷陶瓷膜滤液调节至ph8.0,以分子量2000da的聚醚砜膜进行超滤,以分子量100da的聚醚砜膜进行纳滤后收浓为浓度50mg/ml的浓缩液,将浓缩液调节至ph3.0,加入hp20树脂静态搅拌室温条件下搅拌吸附2h,过筛,水洗干净后树脂装柱,用0.5%磷酸氢二钠
‑
ph10.5溶液和0.75%磷酸氢二钠
‑
ph10.5溶液(各5倍体积)进行梯度洗脱,收集洗脱液,经色谱测试,洗脱液纯度为:94.38%,收率为92.52%。将洗脱液旋转蒸发浓缩至浓度为67mg/ml,调节至ph3.5,60℃下加入活性炭0.3%(质量体积比)搅拌除杂30分钟,过滤,滤液继续旋转蒸发浓缩至浓度为500mg/ml,取出,60℃开始按照每小时降温1℃至40℃后,每20分钟降温1℃速率降温至5℃,分离,得到白色晶体(参见图3),减压干燥(
‑
0.09mpa,60℃,20h),成品经检测,色谱纯度为:99.91%(参见图2,具体各峰检测信息见表2),含量:100.86%,收率为96.45%。最终整体收率为82.75%。
[0053]
表2
[0054]
峰#保留时间面积面积%16.145310602799.91227.2163840.01239.49923580.076总计 3108769100.000
[0055]
实施例3
[0056]
本实施例提供一种本发明的红景天苷分离提取方法,具体如下:
[0057]
将红景天苷陶瓷膜滤液调节至ph8.0,以分子量2000da的聚醚砜膜进行超滤,以分子量100da的聚醚砜膜进行纳滤后收浓为浓度50mg/ml的浓缩液,将浓缩液调节至ph5,加入hp20树脂静态搅拌室温条件下吸附2h,过筛,水洗干净后树脂装柱,用0.5%磷酸氢二钠
‑
ph10.5溶液和0.75%磷酸氢二钠
‑
ph10.5溶液(各5倍体积)进行梯度洗脱,收集洗脱液,经色谱测试,洗脱液纯度:92.03%,收率为93.01%。将洗脱液旋转蒸发浓缩至浓度为70mg/ml,调节至ph3.5,60℃下加入活性炭0.5%(质量体积比)搅拌除杂30分钟,过滤,滤液继续旋转蒸发浓缩至浓度为500mg/ml,取出,60℃开始按照每小时降温1℃至40℃后,每20分钟降温1℃速率降温至5℃,分离,得到白色晶体,减压干燥(
‑
0.09mpa,60℃,20h),成品经检测,色谱纯度为:99.21%,含量:99.07%,收率为93.15%。最终整体收率为80.07%。
[0058]
对比例1
[0059]
本对比例比较不同洗脱液对于红景天苷分离提取效果的影响。具体将实施例2经吸附浓缩液后的树脂搅拌均匀后分成4份,分别以
①
0.25%磷酸氢二钠
‑
ph10.5溶液,
②
0.5%磷酸氢二钠
‑
ph10.5溶液,
③
0.75%磷酸氢二钠
‑
ph10.5溶液,
④
1%磷酸氢二钠
‑
ph10.5溶液进行解吸(洗脱倍数相同,均为5倍),并计算收率。
[0060]
最终以
①
0.25%磷酸氢二钠
‑
ph10.5溶液解吸后的收率为15.78%、纯度为88.75%。以
②
0.5%磷酸氢二钠
‑
ph10.5溶液解吸后的收率为39.57%、纯度为93.52%,以
③
0.75%磷酸氢二钠
‑
ph10.5溶液解吸后的收率为52.78%,纯度:94.39%,以
④
1%磷酸氢二钠
‑
ph10.5溶液解吸后的收率为:89.97%,纯度:91.98%。
[0061]
对比例2
[0062]
本对比例比较活性炭处理时的不同ph值对于红景天苷分离提取效果的影响。具体将实施例2获得的洗脱液旋转蒸发浓缩至浓度为67mg/ml,分别调节至ph3.5和,ph5.0和ph6.0,60℃下加入活性炭0.3%(质量体积比)搅拌除杂30分钟,过滤、计算收率,并将滤液稀释至相同浓度,观察滤液颜色,参见图4。从图4可知,以ph3.5处理效果更好。
[0063]
最终在ph3.5下活性炭处理后的收率为92.6%。在ph5.0下活性炭处理后的收率为89.15%,在ph6.0下活性炭处理后的收率为83.9%。
[0064]
此外,本对比例还对比了以活性炭在ph3.5下处理前后的浓缩液颜色(前后浓度均为500mg/ml)。参见图5。进一步体现了,以ph3.5处理后的脱色效果。
[0065]
对比例3
[0066]
本对比例比较不同降温结晶方式对于红景天苷分离提取效果的影响。具体将实施例2经活性炭处理后的500mg/ml的浓缩液以
①
60℃开始每小时降温3℃的速率降温至5℃方式进行析晶,并计算收率和纯度。以
②
60℃开始每小时降温1℃至50℃,再每小时降温3℃的速率降温至5℃方式进行析晶,并计算收率和纯度。以
③
60℃开始每小时降温2℃至40℃,再每小时降温3℃的速率降温至5℃的方式进行析晶,并计算收率和纯度。以
④
60℃开始每小时降温1℃至40℃,再每小时降温3℃的速率降温至5℃的方式进行析晶,并计算收率和纯度。
[0067]
最终以
①
60℃开始每小时降温3℃的速率降温至5℃方式析晶后的收率为93.77%、纯度为98.15%。以
②
60℃开始每小时降温1℃至50℃,再每小时降温3℃的速率降温至5℃方式析晶后的收率为93.65%、纯度为98.77%。以
③
60℃开始每小时降温2℃至40
℃,再每小时降温3℃的速率降温至5℃的方式进行析晶后的收率为94.05%,纯度为99.01%。
[0068]
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1