一种RXR激动剂的制备方法
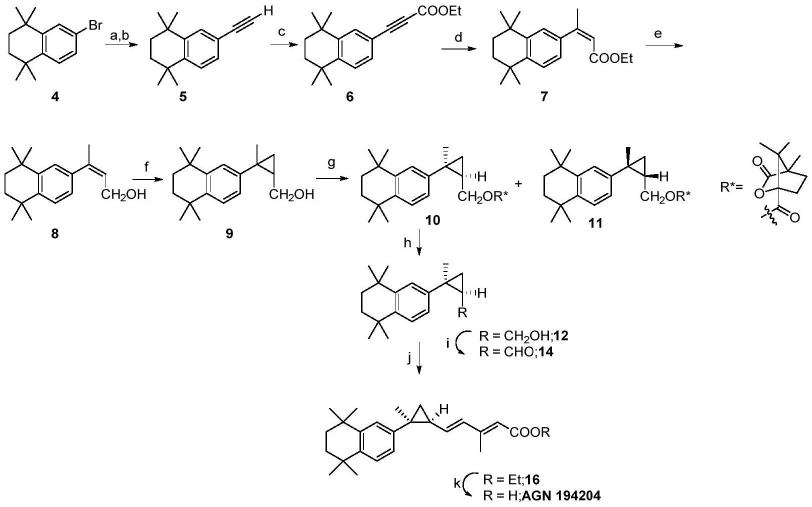
一种rxr激动剂的制备方法
技术领域
1.本发明涉及有机化学合成技术领域,更具体地,涉及一种rxr激动剂的制备方法。
背景技术:
2.rxr激动剂是一种具有口服活性的和选择性的rxr激动剂,对rxrα,rxrβ和rxrγ的kd值分别为0.4nm,3.6nm和3.8nm,ec50分别为0.2nm,0.8nm和0.08nm。agn194204对rar无活性,并具有抗炎和抗癌作用(j.med.chem.2001,44,2298-2303),因此其是一种很重要的rxr激动剂。
3.文献报道可以通过以下路线合成:
[0004][0005]
但该路线步骤长达11步,其中多个步骤反应条件苛刻(例如在-78℃、锂试剂的条件),且需要借助hplc分离异构体,对于工业应用存在较大限制。
技术实现要素:
[0006]
本发明为克服上述合成路线步骤多,反应条件苛刻的缺陷,提供一种rxr激动剂的制备方法。
[0007]
为实现上述目的,本发明采用的技术方案是:
[0008]
一种rxr激动剂的制备方法,包括如下步骤:
[0009]
s1.式a所示化合物与式b所示化合物在氮杂卡宾铜催化剂、提供氢源的硅试剂、溶剂和有机碱的条件下,反应生成式c所示化合物;
[0010]
s2.式c所示化合物与式d所示化合物在荷维达-格鲁布斯ⅱ催化剂条件下反应生成式e所示化合物;
[0011]
s3.式e所示化合物脱除乙基,生成rxr激动剂;
[0012][0013]
其中,r为甲酸甲酯基、乙酰基、对甲苯磺酰基或三氟甲磺酰基。
[0014]
本发明的制备方法反应步骤少,反应条件温和,收率高,能够适用于工业生产制备rxr激动剂。
[0015]
优选地,步骤s1.所述氮杂卡宾铜催化剂选自iprcucl、siprcucl、imescucl或simescucl中的一种。
[0016]
优选地,步骤s1.所述氮杂卡宾铜催化剂为iprcucl。当选择iprcucl作为氮杂卡宾铜催化剂时,其收率更高。
[0017]
优选地,步骤s1.所述提供氢源的硅试剂选自苯硅烷、三苯基硅烷、聚甲基氢硅氧烷、四甲基二硅氮烷、三甲氧基硅烷或三乙基硅烷中的一种。
[0018]
优选地,步骤s1.所述有机碱选自叔丁醇锂、叔丁醇钾或叔丁醇钠。
[0019]
优选地,步骤s1.的反应温度为25~60℃。
[0020]
优选地,步骤s1.所述溶剂为乙二醇二甲醚、四氢呋喃、甲苯、二氯甲烷、二氯乙烷、乙腈或1,4-二氧六环中的一种或多种。
[0021]
优选地,s3.中,式e所示化合物在无机碱作用下脱除乙基。
[0022]
步骤s3.中所述无机碱为常见的促进酯基水解的碱,优选地,步骤s3.所述无机碱选自氢氧化钾和/或氢氧化钠。
[0023]
式a所述化合物可以通过市售购买或者文献公开的技术方案制备得到,优选地,式a所示化合物通过下述反应制备得到:
[0024][0025]
所述保护试剂为氯甲酸甲酯、对甲苯磺酰氯、乙酸酐或三氟甲磺酸酐中的一种。
[0026]
作为一种实施方式,可以按照以下操作制备:取干燥的烧瓶,加入式a1所示化合物和干燥的四氢呋喃,冷却。加入乙炔基溴化镁,升至室温。反应结束后重新冷却,加入氯化铵溶液淬灭。有机相用乙酸乙酯萃取,硫酸钠干燥,减压除去溶剂。将残余物加入二氯甲烷溶解,冷却,加入吡啶,滴入氯甲酸甲酯。滴加完毕后加入4-二甲氨基吡啶,反应升至室温。加入盐酸,有机相用二氯甲烷萃取,硫酸钠干燥,减压除去溶剂后柱层析分离得到式a所示化合物。
[0027]
式b化合物可以通过市售购买或文献公开的技术方案制备得到。
[0028]
优选地,式b所示化合物可根据现有技术(journal of the american chemical society,2013,vol.135,24,p.9083-9090)合成。
[0029]
作为一种实施方式,可以按照以下操作制备:取干燥的烧瓶,加入镁屑。氮气置换后加入干燥的四氢呋喃和6-溴-1,1,4,4-四甲基-1,2,3,4-四氢萘(式b1所示化合物)。室温下搅拌制备得到格式试剂。另取一干燥烧瓶,氮气置换后加入三甲氧基硼和乙醚并冷却。随后将上述制备的格式试剂用注射器逐滴加入至该烧瓶中,继续搅拌直至反应完全。加入稀盐酸后进行后处理得到式b2所示化合物。取一干燥希莱克管,加入2-溴丙烯、醋酸钯、三苯
基膦及磷酸钾并置换氮气。随后加入式b2所示化合物及干燥的甲苯。加热并继续搅拌。反应完成后冷却,转移至分液漏斗后加入水后进行后处理得到式b所示化合物。
[0030][0031]
式d化合物可以通过市售购买或文献公开的技术方案制备得到。
[0032]
优选地,式d所示化合物可根据现有技术(organic letters,2019,vol 21,#1,p.271-274;journal of organic chemistry,2001,vol.66,7,p.2506-2508)合成。
[0033]
作为一种实施方式,可以按照以下操作制备:取乙醛酸乙酯和2-(三苯基膦烯)丙醛于烧瓶中,加入干燥的二氯甲烷。冷却后加入三乙胺。加完后并继续搅拌直至反应完全。减压除去溶剂后柱层析分离得到式d2所示化合物。称取溴代甲基三苯基膦于另一烧瓶中,氮气置换后加入干燥的四氢呋喃并冷却。随后逐滴加入正丁基锂溶液并继续搅拌。另取一干燥烧瓶,氮气置换后将式d2所示化合物置于其中并加入干燥四氢呋喃溶解。冷却后,将上述制备的试剂用注射器逐滴加入该烧瓶中,并继续反应直至tlc监测原料反应完全。加入饱和氯化铵溶液,随后进行后处理得到式d所示化合物。
[0034][0035]
与现有技术相比,本发明的有益效果是:
[0036]
本发明的rxr激动剂agn194204的制备方法反应步骤少,反应条件温和,减少了较危险的锂试剂的使用,大大提升了安全性能,制备得到的agn194204收率高,有望用于大规模的工业生产。
具体实施方式
[0037]
以下结合具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备,可以直接购买或通过已知文献方法合成。
[0038]
实施例1
[0039]
本实施例提供一种rxr激动剂的制备方法,具体如下:
[0040]
首先制备式a所示化合物、式b所示化合物和式d所示化合物:
[0041]
(1)制备式a所示化合物:取干燥的烧瓶,加入式a1所示化合物(500mg,4.1mmol,1.0eq)和干燥的四氢呋喃(20ml),冷却至0℃。加入乙炔基溴化镁(0.5mol/l in thf,12.2ml,6.1mmol,1.5eq),缓慢升至室温。反应结束后重新冷却至0℃,加入氯化铵溶液淬灭。有机相用乙酸乙酯萃取,硫酸钠干燥,减压除去溶剂。将残余物加入二氯甲烷(20ml)溶解,冷却至0℃,加入吡啶(1.0ml,12.2mmol,3.0eq),滴入氯甲酸甲酯(573.4mg,6.1mmol,1.5eq)。滴加完毕后加入4-二甲氨基吡啶(50mg,0.41mmol,0.1eq),反应升至室温保温1小时。加入2n盐酸,有机相用二氯甲烷萃取,硫酸钠干燥,减压除去溶剂后柱层析分离(石油
醚/乙酸乙酯=20/1)得到式a所示化合物(676.6mg,80%)。
[0042]1h nmr(500mhz,cdcl3)δ5.18(td,j=6.5,2.2hz,1h),3.77(s,3h),3.51(t,j=6.6hz,2h),2.51(d,j=2.2hz,1h),1.86
–
1.76(m,4h),1.65
–
1.54(m,2h).
13
cnmr(126mhz,cdcl3)δ154.9,80.2,74.8,67.5,55.0,44.5,33.8,31.9,22.2.ir(kbr,cm-1
)3292,2957,1752,1443,1266,958,790,650.hrms(esi)calcd for c9h
14
clo
3+
[m+h]
+
:205.0626,found:205.0634.
[0043]
(2)制备式b所示化合物:取一干燥的带搅拌子的100ml烧瓶,加入镁屑(360mg,2.0eq)。氮气置换后加入20ml干燥的四氢呋喃和式b1所示化合物(2.0g)。室温下搅拌3小时制备得到格式试剂。另取一带搅拌子的100ml干燥烧瓶,氮气置换后加入三甲氧基硼(1.2g,1.5eq)和10ml乙醚并冷却至0℃。随后将上述制备的格式试剂用注射器逐滴加入至该烧瓶中,继续搅拌1小时直至反应完全。加入稀盐酸后用乙醚萃取3次,干燥,减压除去溶剂,柱层析得到式b2所示化合物(1.3g,75%)。取一带搅拌的50ml干燥希莱克管,加入2-溴丙烯(1.3g,2.0eq)、醋酸钯(125mg,10%)、三苯基膦(294mg,20%)及磷酸钾(3.56g,3.0eq)并置换氮气。随后加入式b2所示化合物(1.3g)及15ml干燥的甲苯。加热至90℃并继续搅拌5小时。反应完成后冷却至室温,转移至分液漏斗后加入水并用乙酸乙酯萃取3次,干燥,减压除去溶剂后柱层析(洗脱剂:石油醚)分离得到式b化合物(1.1g,86%)。
[0044]
(3)制备式d所示化合物:取乙醛酸乙酯(d1化合物,1.0g)和2-(三苯基膦烯)丙醛(4.6g,1.5eq.)于带搅拌的100ml烧瓶中,加入50ml干燥的二氯甲烷。冷却至0℃后逐滴加入三乙胺(2.7ml,2.0eq)。加完后缓慢升至室温并继续搅拌3小时直至反应完全。减压除去溶剂,柱层析(洗脱剂:石油醚/乙酸乙酯=10/1)分离得到d2所示化合物(1.1g,79%)。称取溴代甲基三苯基膦(4.1g,1.5eq)于带搅拌的50ml烧瓶中,氮气置换后加入20ml干燥的四氢呋喃并冷却至0℃。随后逐滴加入正丁基锂溶液(4.6ml,1.5eq,2.5m)并继续搅拌1小时。另取一带搅拌的100ml干燥烧瓶,氮气置换后将d2所示化合物(1.1g)置于其中并加入10ml干燥四氢呋喃。冷却至0℃后,将上述制备的试剂用注射器逐滴加入该烧瓶中,并继续反应2小时直至tlc监测原料反应完全。加入饱和氯化铵溶液,随后用乙醚萃取3次,干燥,减压除去多余溶剂,柱层析分离得到式d所示化合物(0.9g,83%)。
[0045]
rxr激动剂agn194204的制备方法,包括如下步骤:
[0046]
s1.制备式c所示化合物:取一干燥的带搅拌子的希莱克管加入叔丁醇锂(78mg,0.98mmol,1.0eq)、iprcucl(4.8mg,0.98%mmol,1%eq)和三苯基硅烷(291mg,1.12mmol,1.2eq),氮气置换三次后加入乙二醇二甲醚(10ml),室温搅拌5分钟。加入式a所示化合物(200mg,0.98mmol,1.0eq)的乙二醇二甲醚溶液(1ml),继续搅拌5分钟。加入式b所示化合物(342mg,1.5mmol,1.5eq),升至40℃保温12小时。加入饱和氯化铵溶液,随后用乙酸乙酯萃取(3
×
10ml),有机相饱和食盐水洗,硫酸钠干燥,减压除去多余溶剂,柱层析分离(洗脱剂:石油醚)得到式c所示化合物(208mg,59%)。
[0047]
s2.制备式e所示化合物:取一干燥的带搅拌子的希莱克管加入式c所示化合物(100mg,0.28mmol,1.0eq),氮气置换三次后加入二氯甲烷(10ml)、式d所示化合物(118mg,0.84mmol,3.0eq)、hoveyda
–
grubbs ii催化剂(17mg,0.028mmol,0.1eq)。反应加热回流24小时。tlc监控原料反应完全后,冷却至室温。减压除去多余溶剂,柱层析分离(洗脱剂石油醚/乙酸乙酯=20/1)得到式e所示化合物(81mg,76%)。
[0048]
s3.制备agn 194204:取一干燥的带搅拌子的烧瓶加入式e所示化合物(60mg,0.16mmol,1.0eq)、四氢呋喃2ml、甲醇2ml、氢氧化钠水溶液(0.4ml,2n,5.0eq),40℃搅拌3小时至原料反应完全。冷却至室温后加入盐酸(1ml,2n)调节ph=6。随后用乙酸乙酯萃取(3
×
10ml),有机相饱和食盐水洗,硫酸钠干燥,减压除去多余溶剂,柱层析分离(石油醚/乙酸乙酯=3/1)得到agn 194204(46mg,81%)。
[0049]1h nmr(500mhz,cdcl3)δ7.21(d,j=8.1hz,1h),7.12(d,j=2.0hz,1h),7.02(dd,j=8.1,2.0hz,1h),6.20(d,j=15.5hz,1h),5.64(s,1h),5.26(dd,j=15.5,10.0hz,1h),1.97(d,j=1.1hz,3h),1.72(ddd,j=10.1,8.2,5.1hz,1h),1.66(d,j=1.6hz,4h),1.42(s,3h),1.26(s,9h),1.20(s,3h),1.19
–
1.16(m,1h),1.14(t,j=5.0hz,1h).
13
c nmr(126mhz,cdcl3)δ172.5,155.2,144.6,142.9,141.5,139.6,131.3,127.6,126.5,126.3,115.3,35.2,35.1,34.2,34.0,32.0,31.9,31.8,30.7,29.8,28.7,22.2,13.7.ir(kbr,cm-1
)2959,2919,1664,1600,1442,1240,1075,959,819,440.hrms(esi)calcd for c
24h32
nao
2+
[m+na]
+
:375.2295,found:375.2299.
[0050][0051]
实施例2~9
[0052]
实施例2~9提供一系列rxr激动剂,其制备方法与实施例1相同,其区别在于,采用不同氮杂卡宾铜催化剂以及不同含量的氮杂卡宾铜催化剂,具体见表1,收率采用步骤s1的式c所示化合物收率表示。
[0053]
表1实施例2~9
[0054]
实施例催化剂式c所示化合物收率/%2siprcucl(0.1%)503imescucl(0.1%)344simescucl(0.1%)295iprcucl(10%)606iprcucl(2%)597iprcucl(1%)598iprcucl(0.5%)579iprcucl(0.01%)20
[0055]
从实施例1~9看,当氮杂卡宾铜催化剂的含量为0.01%eq还具有20%的收率,当iprcucl作为氮杂卡宾铜催化剂时具有较好的收率。
[0056]
实施例10~16
[0057]
实施例10~16提供一系列rxr激动剂,其制备方法与实施例1相同,其区别在于,采用不同提供氢源的硅试剂及不同含量的硅试剂制备得到,具体见表2,采用步骤s1的式c所示化合物收率表示。
[0058]
表2实施例10~16
[0059][0060][0061]
从实施例10~14看,苯硅烷、三苯基硅烷、聚甲基氢硅氧烷、四甲基二硅氮烷、三甲氧基硅烷、三乙基硅烷都能制备得到式c所示化合物。
[0062]
实施例17~19
[0063]
实施例17~19提供一系列rxr激动剂,其制备方法与实施例1相同,其区别在于,采用不同r基的式a所示化合物制备,具体见表3,收率采用步骤s1的式c所示化合物收率表示。
[0064]
表3实施例17~19
[0065][0066]
实施例20~22
[0067]
实施例20~22提供一系列rxr激动剂,其制备方法与实施例1相同,其区别在于,步骤s1或s3中的碱不相同,具体见表4。
[0068]
表4实施例20~22
[0069][0070]
实施例23~32
[0071]
实施例23~32提供一系列rxr激动剂,其制备方法与实施例1相同,其区别在于,步骤s1采用了不同的反应溶剂、温度和时间,具体见表5,收率用步骤s1中的式c所示化合物收率表示。
[0072]
表5实施例23~32
[0073][0074][0075]
从实施例1和23~24看,当温度在25~60℃都具有较好的收率。
[0076]
从实施例1和25~26看,当反应时间在6~24h时具有较好的收率。
[0077]
从实施例1和27~32看,采用不同的溶剂都能制备得到rxr激动剂。
[0078]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1