一种砜吡草唑农药中间体的合成方法与流程
1.本发明涉及的是化学合成的技术领域,尤其涉及的是一种砜吡草唑农药中间体的合成方法。
背景技术:
2.5,5
‑
二甲基异恶唑化合物是一种重要的农药中间体,可以用于砜吡草唑等农药的合成。目前合成该化合物的主要方法为异丁烯与二卤代甲醛肟的成环反应制备或者是使用较为复杂的催化体系。
3.在专利文献(wo2006068092a1),公开了一种制备3
‑
卤代
‑
5,5
‑
二甲基异恶唑化合物的方法。该方法以二溴甲醛肟与异丁烯在醋酸丁酯的条件先进行成环生成5,5
‑
二甲基异恶唑。反应通式如下:
[0004][0005]
该方法在制备3
‑
卤代
‑
5,5
‑
二甲基异恶唑化合物时,使用了比较危险的异丁烯气体,在大规模工业化生产中存在安全隐患;该方法还使用了醋酸正丁酯作为溶剂,该溶剂沸点较高,而且在碱性条件下会水解,难以多次回收套用,会加大后处理难度和生产成本。
[0006]
在专利文献(wo2008000469),公开了一种制备3
‑
卤代
‑
5,5
‑
二甲基异恶唑化合物的方法。该方法以丙酮肟和3
‑
甲基
‑2‑
丁烯醛为原料,以有机碱和有机酸作为混合催化剂,反应40h以上得到5,5
‑
二甲基异恶唑化合物,然后与氯气反应得到3
‑
氯
‑
5,5
‑
二甲基异恶唑化合物。反应通式如下:
[0007][0008]
该方法在制备3
‑
卤代
‑
5,5
‑
二甲基异恶唑化合物时,首先,该反应使用了有机酸碱催化剂,如n
‑
甲基苯胺和三氟乙酸,该催化剂难以回收套用,同时对回收溶剂会造成一定影响,并且该催化剂还会增加生产成本;其次,该反应使用了氯气作为氯化试剂,在工业化生产中也会存在安全隐患。
[0009]
基于上述内容,提出一种砜吡草唑农药中间体的合成方法。
技术实现要素:
[0010]
本发明的目的在于克服现有技术的不足,提供了一种砜吡草唑农药中间体的合成方法,以解决现有方法总收率较低的技术问题。
[0011]
本发明是通过以下技术方案实现的:
[0012]
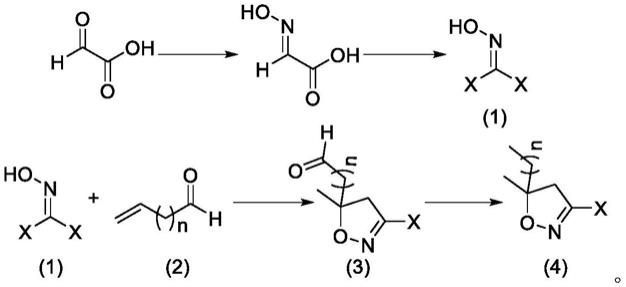
一种砜吡草唑农药中间体的合成方法,所述砜吡草唑农药中间体为3
‑
卤代
‑
5,5
‑
二甲基异恶唑化合物,化学结构为通式(4)所示,该合成方法乙醛酸为起始原料,包括以下步骤:
[0013]
(1)乙醛酸经过成肟反应获得乙醛肟甲酸;
[0014]
(2)所述乙醛肟甲酸经过卤化反应获得通式(1)所示化合物;
[0015]
(3)所述通式(1)所示化合物与通式(2)所示化合物进行[3+2]环化反应获得异恶唑环;
[0016]
(4)所述异恶唑环通过黄鸣龙反应获得通式(4)所示化合物,即3
‑
卤代
‑
5,5
‑
二甲基异恶唑。
[0017]
其中,(1)至(4)反应通式如下:
[0018][0019]
作为对上述发明的进一步改进,所述通式(2)至(4)所示化合物中n=0
‑
1。
[0020]
作为对上述发明的进一步改进,所述步骤(2)中卤化反应或步骤(3)中环化反应使用溶剂为氯代烷烃类溶剂、醇类溶剂、酯类溶剂中的一种,优选为氯代烷烃类溶剂。
[0021]
作为对上述发明的进一步改进,所述氯代烷烃类溶剂为二氯甲烷、二氯乙烷、乙腈、四氯化碳、氯仿中的一种或多种。
[0022]
作为对上述发明的进一步改进,所述醇类溶剂为甲醇、乙醇中的一种。
[0023]
作为对上述发明的进一步改进,所述酯类溶剂为乙酸乙酯、乙酸丁酯中的一种。
[0024]
作为对上述发明的进一步改进,所述步骤(4)中黄鸣龙反应使用溶剂为缩醇类溶剂。
[0025]
作为对上述发明的进一步改进,所述缩醇类溶剂为二缩乙二醇、三缩乙二醇中的一种。
[0026]
作为对上述发明的进一步改进,所述步骤(2)中卤化反应、步骤(3)中环化反应或步骤(4)中黄鸣龙反应使用的碱为无机碱、有机碱中的一种,优选用无机碱。
[0027]
作为对上述发明的进一步改进,所述无机碱为碳酸钾、碳酸钠、氢氧化钾、氢氧化钠中的一种,优选为氢氧化钾、氢氧化钠。
[0028]
作为对上述发明的进一步改进,所述有机碱为三乙胺、吡啶、甲醇钠、甲醇钾中的一种。
[0029]
作为对上述发明的进一步改进,所述步骤(2)中合成通式(1)所示化合物具体步骤包括:在
‑
20
‑
50℃下,将乙醛肟甲酸与有机溶剂在釜中混合,然后向釜中加入碱,搅拌后缓慢加入卤化试剂,在10
‑
100℃下保温反应;反应完成后,用水洗涤2次分出有机相后除去溶剂,获得通式(1)所示化合物。
[0030]
作为对上述发明的进一步改进,所述卤化试剂为溴、氯气、五氯化磷加溴化氢加双
氧水、n
‑
溴代丁二酰亚胺加双氧水、三氯氧磷中的一种。
[0031]
作为对上述发明的进一步改进,所述乙醛肟甲酸与卤化试剂的摩尔比为1:1
‑
10,乙醛肟甲酸与碱的摩尔比为1:1
‑
10,乙醛肟甲酸与溶剂的质量比为1:1
‑
100。
[0032]
作为对上述发明的进一步改进,所述卤化试剂滴加速度为0.01
‑
1h/kg,反应保温时间为0.1
‑
10h。
[0033]
作为对上述发明的进一步改进,所述步骤(3)中合成通式(3)所示化合物具体步骤包括:在
‑
20
‑
50℃氮气保护条件下,将通式(1)所示化合物与溶剂混合后向反应中加入碱,再向反应体系中滴加通式(2)所示化合物;将反应在
‑
20
‑
50℃条件下保温直至反应完全;反应结束后,将反应产物过滤,再用水洗涤有机相,将有机相进行蒸馏得到通式(3)所示化合物。
[0034]
作为对上述发明的进一步改进,所述通式(1)所示化合物与溶剂的质量比为1:0.1
‑
20,通式(1)所示化合物与碱的摩尔比为1:0.1
‑
20,通式(1)所示化合物与通式(2)所示化合物的摩尔比为1:0.1
‑
5。
[0035]
作为对上述发明的进一步改进,所述保温时间为0.1
‑
20h。
[0036]
作为对上述发明的进一步改进,所述步骤(4)中合成通式(4)所示化合物步骤包括:将通式(3)所示化合物与溶剂混合后和体积比含量为1%
‑
100%水合肼混合,然后加热到100
‑
200℃,保持该温度直至通式(3)所示化合物转化完全,然后降至常温后向反应中加入碱,随后继续将反应升温至100
‑
250℃,保温直至反应结束,反应结束后降温至室温,然后加入酸将反应中和,之后用溶剂对反应进行萃取,将得到的溶剂用水洗涤三次之后浓缩,减压蒸馏得到通式(4)所示化合物。
[0037]
作为对上述发明的进一步改进,所述通式(3)所示化合物与溶剂的质量比为1:1
‑
20,通式(3)所示化合物与水合肼的摩尔比为1:0.5
‑
50,通式(3)所示化合物与碱的摩尔比1:1
‑
20。
[0038]
本发明相比现有技术具有以下优点:本发明提供了一种砜吡草唑农药中间体的合成方法,砜吡草唑农药中间体为3
‑
卤代
‑
5,5
‑
二甲基异恶唑化合物,属于化学合成的技术领域,该方法以乙醛酸为原料,经过成肟反应、卤化反应、环化反应和还原反应得到3
‑
卤代
‑
5,5
‑
二甲基异恶唑化合物,相比现有报道的合成技术相比,起始原料简单易得,不使用危险原料和危险工艺,且工艺操作简单,不需要纯化可以直接进行下一步反应,减少了三废的产生,得到的3
‑
卤代
‑
5,5
‑
二甲基异恶唑化合物的含量在95%以上。该发明可应用于工业生产,对降低生产成本和提高产品品质有重要的意义。
具体实施方式
[0039]
本发明提供了一种砜吡草唑农药中间体的合成方法,所述砜吡草唑农药中间体为3
‑
卤代
‑
5,5
‑
二甲基异恶唑化合物,化学结构为通式(4)所示,该合成方法乙醛酸为起始原料,包括以下步骤:
[0040]
(1)乙醛酸经过成肟反应获得乙醛肟甲酸;
[0041]
(2)所述乙醛肟甲酸经过卤化反应获得通式(1)所示化合物;
[0042]
(3)所述通式(1)所示化合物与通式(2)所示化合物进行[3+2]环化反应获得异恶唑环;
[0043]
(4)所述异恶唑环通过黄鸣龙反应获得通式(4)所示化合物,即3
‑
卤代
‑
5,5
‑
二甲基异恶唑;
[0044]
其中,(1)至(4)反应通式如下:
[0045][0046]
其中,乙醛肟甲酸的合成、通式(1)所示化合物的合成步骤为常规步骤,可参考专利文献(wo2006068092a1),通式(2)至(4)所示化合物中n=0
‑
1。
[0047]
步骤(2)中合成通式(1)所示化合物具体步骤包括:在
‑
20
‑
50℃下,将乙醛肟甲酸与有机溶剂在釜中混合,然后向釜中加入碱,搅拌后以0.01
‑
1h/kg的卤化试剂滴加速度缓慢加入卤化试剂,在10
‑
100℃下保温反应0.1
‑
10h;反应完成后,用水洗涤2次分出有机相后除去溶剂,获得通式(1)所示化合物。
[0048]
卤化试剂为溴、氯气、五氯化磷加溴化氢加双氧水、n
‑
溴代丁二酰亚胺加双氧水、三氯氧磷中的一种;乙醛肟甲酸与卤化试剂的摩尔比为1:1
‑
10,乙醛肟甲酸与碱的摩尔比为1:1
‑
10,乙醛肟甲酸与溶剂的质量比为1:1
‑
100。
[0049]
步骤(3)中合成通式(3)所示化合物具体步骤包括:在
‑
20
‑
50℃氮气保护条件下,将通式(1)所示化合物与溶剂混合后向反应中加入碱,再向反应体系中滴加通式(2)所示化合物;将反应在
‑
20
‑
50℃条件下保温0.1
‑
20h直至反应完全;反应结束后,将反应产物过滤,再用水洗涤有机相,将有机相进行蒸馏得到通式(3)所示化合物;
[0050]
通式(1)所示化合物与溶剂的质量比为1:0.1
‑
20,通式(1)所示化合物与碱的摩尔比为1:0.1
‑
20,通式(1)所示化合物与通式(2)所示化合物的摩尔比为1:0.1
‑
5。
[0051]
步骤(4)中合成通式(4)所示化合物步骤包括:将通式(3)所示化合物与溶剂混合后和含量为1%
‑
100%水合肼混合,然后加热到100
‑
200℃,保持该温度直至通式(3)所示化合物转化完全,然后降至常温后向反应中加入碱,随后继续将反应升温至100
‑
250℃,保温直至反应结束,反应结束后降温至室温,然后加入酸将反应中和,之后用溶剂对反应进行萃取,将得到的溶剂用水洗涤三次之后浓缩,减压蒸馏得到通式(4)所示化合物;通式(3)所示化合物与溶剂的质量比为1:1
‑
20,通式(3)所示化合物与水合肼的摩尔比为1:0.5
‑
50,通式(3)所示化合物与碱的摩尔比1:1
‑
20。
[0052]
步骤(2)中卤化反应或步骤(3)中环化反应使用溶剂为氯代烷烃类溶剂、醇类溶剂、酯类溶剂中的一种,优选为氯代烷烃类溶剂,氯代烷烃类溶剂具体为二氯甲烷、二氯乙烷、乙腈、四氯化碳、氯仿中的一种或多种,醇类溶剂具体为甲醇、乙醇中的一种,酯类溶剂具体为乙酸乙酯、乙酸丁酯中的一种。
[0053]
步骤(4)中黄鸣龙反应使用溶剂为缩醇类溶剂,具体为二缩乙二醇、三缩乙二醇中的一种。
[0054]
步骤(2)
‑
(4)中,使用的碱为无机碱、有机碱中的一种,优选用无机碱,无机碱为碳酸钾、碳酸钠、氢氧化钾、氢氧化钠中的一种,优选为氢氧化钾、氢氧化钠,有机碱为三乙胺、吡啶、甲醇钠、甲醇钾中的一种。
[0055]
为了进一步验证本实施的效果,提供以下具体的一种砜吡草唑农药中间体的合成方法的具体实施方式。
[0056]
实施例1
[0057]
(1)二溴甲醛肟的合成
[0058]
向1000ml四口瓶中加入200g含量50%的乙醛酸水溶液,然后加入100g盐酸羟胺和200g水,在25℃下搅拌3h;随后向反应中加入200g二氯甲烷,并且将反应温度降至0℃,然后向反应中滴加250g液溴,滴完之后将反应温度升至25℃,保温3h;反应结束之后将反应分相,将得到的有机相用水洗涤(200ml
×
3),最后将有机相进行减压蒸馏,得到200g二溴甲醛肟淡黄色固体,产率:74%。
[0059]
(2)3
‑
溴
‑5‑
甲基
‑5‑
甲酰基
‑
4,5二氢异恶唑的合成
[0060]
在n2条件下,向1000ml四口瓶中加入200g二溴甲醛肟和300g甲醇和50g水,然后加入148g的khco3(1.5eq),将反应温度控制在0℃;然后向反应中滴加66g丙烯醛(1.2eq),滴加结束后,将反应温度升至30℃,保温2h;反应结束后,向反应中加入500g水,然后用二氯甲烷萃取(300ml
×
3),将有机相合并之后减压蒸馏,得到3
‑
溴
‑5‑
甲基
‑5‑
甲酰基
‑
4,5二氢异恶唑淡黄色液体,重量:170g,含量:95%,产率:94%。
[0061]
(3)3
‑
溴
‑
5,5
‑
二甲基
‑
4,5
‑
二氢异恶唑的合成
[0062]
在n2条件下,向2000ml四口瓶中分别加入170g的3
‑
溴
‑5‑
甲基
‑5‑
甲酰基
‑
4,5二氢异恶唑、300g二乙二醇和235g的koh(5eq);然后向反应中滴加420g水合肼(50%水溶液,5eq),随后将反应温度升至160℃并保温2h;反应结束之后将反应温度降至常温,用hcl(10%)调解反应ph至7
‑
9之间,然后用二氯乙烷萃取(500ml
×
3),最后将有机相合并之后进行减压蒸馏,得到138g的3
‑
溴
‑
5,5
‑
二甲基
‑
4,5
‑
二氢异恶唑淡黄色液体,含量:98%,产率:91%。
[0063]
实施例2
[0064]
(1)二溴甲醛肟的合成
[0065]
向1000ml四口瓶中加入200g含量50%的乙醛酸水溶液,然后加入100g盐酸羟胺和200g水,在25℃下搅拌3h;随后向反应中加入200g二氯甲烷,并且将反应温度降至0℃,然后向反应中通入400g氯气,之后将反应温度升至25℃,保温3h;反应结束之后将反应分相,将得到的有机相用水洗涤(200ml
×
3),最后将有机相进行减压蒸馏,得到132g二氯甲醛肟淡黄色固体,产率:86%。
[0066]
(2)3
‑
溴
‑5‑
甲基
‑5‑
甲酰基
‑
4,5二氢异恶唑的合成
[0067]
在n2条件下,向1000ml四口瓶中加入132g二氯甲醛肟和200g甲醇和50g水,然后加入174g的khco3,将反应温度控制在0℃;然后向反应中滴加62g丙烯醛(1.3eq),滴加结束后,将反应温度升至30℃,保温2h;反应结束后,向反应中加入400g水,然后用二氯甲烷萃取(200ml
×
3),将有机相合并之后减压蒸馏,得到123g的3
‑
氯
‑5‑
甲基
‑5‑
甲酰基
‑
4,5二氢异恶唑淡黄色液体,含量:92%,产率:90%。
[0068]
(3)3
‑
氯
‑
5,5
‑
二甲基
‑
4,5
‑
二氢异恶唑的合成
[0069]
在n2条件下,向500ml四口瓶中分别加入132g的3
‑
氯
‑5‑
甲基
‑5‑
甲酰基
‑
4,5二氢异恶唑、200g二乙二醇和216的koh(5eq);然后向反应中滴加385g水合肼(5eq),随后将反应温度升至160℃保温2h;反应结束之后将反应温度降至常温,用hcl(10%)调解反应ph至7
‑
9之间,然后用二氯乙烷萃取(300ml
×
3),最后将有机相合并之后进行减压蒸馏,得到96g的3
‑
氯
‑
5,5
‑
二甲基
‑
4,5
‑
二氢异恶唑淡黄色液体,含量:98%,产率:92%。
[0070]
实施例3
[0071]
(1)二溴甲醛肟的合成
[0072]
向1000ml四口瓶中加入200g含量50%的乙醛酸水溶液,然后加入100g盐酸羟胺和200g水,在25℃下搅拌3h;随后向反应中加入200g二氯甲烷,并且将反应温度降至0℃,然后向反应中滴加250g液溴,滴完之后将反应温度升至25℃,保温3h;反应结束之后将反应分相,将得到的有机相用水洗涤(200ml
×
3),最后将有机相进行减压蒸馏,得到200g二溴甲醛肟淡黄色固体,产率:74%。
[0073]
(2)3
‑
溴
‑5‑
乙酰基
‑5‑
甲基
‑
4,5二氢异恶唑的合成
[0074]
在n2条件下,向1000ml四口瓶中加入200g二溴甲醛肟和300g甲醇和50g水,然后加入148gkhco3,将反应温度控制在0℃;然后向反应中滴加113g的3
‑
丁烯醛(1.3eq),滴加结束后,将反应温度升至30℃,保温2h;反应结束后,向反应中加入500g水,然后用二氯甲烷萃取(300ml
×
3),将有机相合并之后减压蒸馏,得到171g3
‑
溴
‑5‑
乙酰基
‑5‑
甲基
‑
4,5二氢异恶唑淡黄色液体,含量:95%,产率:79%。
[0075]
(3)3
‑
溴
‑5‑
乙基
‑5‑
甲基
‑
4,5
‑
二氢异恶唑的合成
[0076]
在n2条件下,向500ml四口瓶中分别加入171g3
‑
溴
‑5‑
甲基
‑5‑
甲酰基
‑
4,5二氢异恶唑、300g二乙二醇和400gkoh(5eq);然后向反应中滴加400g水合肼(5eq),随后将反应温度升至160℃保温2h;反应结束之后将反应温度降至常温,用hcl(10%)调解反应ph至7
‑
9之间,然后用二氯乙烷萃取(300ml
×
3),最后将有机相合并之后进行减压蒸馏,得到131g3
‑
溴
‑5‑
乙基
‑5‑
甲基
‑
4,5
‑
二氢异恶唑淡黄色液体,含量:98%,产率:93%。
[0077]
以上为本发明几种详细的实施方式和具体的操作过程,是以本发明技术方案为前提下进行实施,但本发明的保护范围不限于上述的实施例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1