一种基于CRISPR/Cas系统的新型冠状病毒双靶标快速检测方法及试剂盒
一种基于crispr/cas系统的新型冠状病毒双靶标快速检测方法及试剂盒
技术领域
1.本发明属于生物检测技术领域,涉及一种基于crispr/cas系统的新型冠状病毒双靶标快速检测方法及试剂盒。
背景技术:
2.新型冠状病毒(covid
‑
19),简称“新冠病毒”,是一种新发现的单链rna病毒,全长29903个核苷酸,通过飞沫经呼吸道与结膜传播,传染性强,传播范围广,是目前已知的第七种对人类致病的冠状病毒。相比于其它冠状病毒造成的急性症状,新型冠状病毒感染症状从轻微、咳嗽、发烧到危重均有出现,感染症状和常见呼吸系统疾病相似,隐蔽性强,传染性极强。
3.目前针对新冠病毒尚无有效的治疗药物,新冠疫苗的研发需要较长的周期,研发后的疫苗进行全民接种并形成免疫力也需要巨大的疫苗产能以及极长的接种周期。所以防控新冠病毒的最有效手段依然是早检出早隔离,通过核酸检测发现新冠病毒感染者,并对感染者以及相关密切接触者进行隔离从而阻断病毒传播。新冠病毒的核酸诊断金标准是实时荧光定量pcr法(qpcr),该方法通过pcr扩增新冠核酸目标片段,在扩增时每产生一个目标片段即切割断裂一个taq
‑
man探针,产生荧光信号。荧光信号的产生与扩增产物相同步。由于pcr扩增时模板呈指数增长,其达到设置阈值时进行的循环数(ct)与初始模板量的倒数存在对数关系,可据此进行定量分析,从而实现对新冠病毒的定量检测。但是qpcr需要昂贵的热循环仪器以及熟练的操作人员,且检测周期一般较长需要3
‑
4小时,难以满足在一些特殊场景的筛查需求,如资源匮乏地区如偏远农村的疫情防控。
4.现有的一些核酸快速检测方法由rt
‑
lamp(逆转录
‑
环介导等温扩增)以及rt
‑
rpa
‑
exo探针法(逆转录
‑
重组酶聚合酶扩增exo探针法),这类方法通过牺牲部分精确度与灵敏度摆脱了对变温设备的依赖,在一些特殊恒温核酸扩增酶介导下,目的核酸可以在固定温度进行核酸扩增与检测。基于rt
‑
lamp或rt
‑
raa的核酸恒温扩增检测,虽然摆脱了热循环设备的限制,但是检测精确度与灵敏度相比于qpcr则有一个巨大的下降。基于crispr/cas系统的恒温检测则在恒温扩增方法的基础上引入了精确灵敏的crispr系统,极大提高了检测方法的精确度与灵敏度。
5.crispr是“clustered regularly interspaced short palindromic repeats”缩写,指规律成簇的间隔短回文重复。cas是“crispr
‑
associated”缩写,为crispr相关。crispr/cas系统是一个细菌及古细菌进化出来用以抵御噬菌体入侵的适应性机制,后被发现并发展成为一种由引导rna指引cas核酸酶对靶向基因进行特定核酸编辑的技术。crispr/cas系统的工作原理是crrna(crispr
‑
derived rna)通过碱基配对与tracrrna(trans
‑
activating rna)结合形成tracrrna/crrna复合物,此复合物引导核酸酶如cas9蛋白至与crrna配对的序列靶位点处剪切双链dna,从而实现对基因组dna序列进行编辑;而通过人工设计这两种rna,可以改造形成具有引导作用的grna(guide rna),足以引导cas9对
dna的定点切割。目前,基于crispr的核酸快速检测技术主要分为两大类,即2020年诺贝尔化学奖得主jennifer doudna开发的依赖于cas12a的称为detectr的核酸恒温检测技术,以及crispr专利所有者张锋开发的依赖于cas13a的称为sherlock的核酸恒温检测技术。其原理都是通过恒温扩增方法如rpa对目的片段进行扩增富集,扩增后的目的片段会被cas蛋白通过一段crrna的引导靶向识别,激活cas蛋白成为一个dna切割机(cas12a)或者rna切割机(cas13a),将附近所有的单链dna(cas12a)或者单链rna(cas13a)进行切割。这一特性作用于单链核酸荧光探针则可用于报告检测结果。
6.detectr系统以及sherlock系统一次只能检测单一位点,无法像qpcr一样通过添加不同靶位点的taq
‑
man探针进行双靶标甚至三靶标的检测。在遇到一些低拷贝的临床样本时,多重靶标的检测方法可以避免因为新冠病毒核酸基因组不全导致的假阴性。而基于crispr/cas系统的detectr以及sherlock系统目前都只能对单一靶标进行检测。但实验发现每种不同的cas蛋白被激活后仅能切割对应的单链dna探针(lbacas12a)或单链rna探针(lwacas13a),这使得将两种crispr系统整合为单一体系进行双靶标的同时检测成为可能。
技术实现要素:
7.本发明的目的在于提供一种基于crispr/cas系统的新型冠状病毒双靶标快速检测方法及试剂盒。
8.为实现上述目的及其他相关目的,本发明提供的技术方案是:一种基于crispr/cas系统的新型冠状病毒双靶标快速检测的试剂盒,所述试剂盒包括:
9.两组rt
‑
raa扩增引物:
10.cas13
‑
s
‑
for:5
’‑
gaaattaatacgactcactataggggctatcatcttatgtccttccctcagtcag
‑3’
;
11.cas13
‑
s
‑
rev:5
’‑
aatggcaggagcagttgtgaagttcttttc
‑3’
;
12.cas12
‑
n
‑
for:5
’‑
aatgtcgcgcattggcatggaagtcaca
‑3’
;
13.cas12
‑
n
‑
rev:5
’‑
gacttgatctttgaaatttggatctttg
‑3’
;
14.两种crispr特异性检测的crrna:
15.cas12a
‑
n
‑
crrna:
[0016]5’‑
uaauuucuacuaaguguagauauggcaccuguguaggucaa
‑3’
;
[0017]
cas13a
‑
s
‑
crrna:
[0018]5’‑
gauuuagacuaccccaaaaacgaaggggacuaaaaccauaagucacaugcaagaagacuacacc
‑3’
;
[0019]
rna荧光探针:5
’‑
fam
‑
mararurgrgrcmamararurgrgrcma
‑
bhq1
‑3’
;
[0020]
其中,m代表2位氧甲基修饰,r代表核糖核苷酸;
[0021]
dna荧光探针:5
’‑
vic
‑
ttattatt
‑
bhq1
‑3’
。
[0022]
优选的技术方案为:还包括buffer a溶液和bufferb溶液;buffer a溶液配置方法为:向1l水中加入50mmol的tris缓冲液,100nmol的醋酸钾,20g的聚乙二醇粉末和2mmol二硫苏糖醇;bufferb溶液为浓度为280mm的醋酸镁溶液。
[0023]
优选的技术方案为:还包括:hepes缓冲液、mgcl2溶液、10
×
neb buffer2.1缓冲液、rnase inhibitor、t7 rna polymerase、无rna酶水。
[0024]
为实现上述目的及其他相关目的,本发明提供的技术方案是:一种基于crispr/cas系统的新型冠状病毒双靶标快速检测方法,包括下列步骤:
[0025]
步骤1:将待检测样本浸入病毒保存液中,然后用rna提取试剂盒进行核酸提取,得到待检核酸;
[0026]
步骤2:向一容纳有蛋白酶冻干粉的反应管中,加入37.5μl的buffer a溶液,2μl的cas13
‑
s
‑
for,2μl的cas13
‑
s
‑
rev,2μl的cas12
‑
n
‑
for,2μl的cas12
‑
n
‑
rev以及2μl待检核酸,然后加2.5μl的bufferb溶液,盖上反应管的管盖后进行扩增反应,得到核酸扩增产物;
[0027]
cas13
‑
s
‑
for:5
’‑
gaaattaatacgactcactataggggctatcatcttatgtccttccctcagtcag
‑3’
;
[0028]
cas13
‑
s
‑
rev:5
’‑
aatggcaggagcagttgtgaagttcttttc
‑3’
;
[0029]
cas12
‑
n
‑
for:5
’‑
aatgtcgcgcattggcatggaagtcaca
‑3’
;
[0030]
cas12
‑
n
‑
rev:5
’‑
gacttgatctttgaaatttggatctttg
‑3’
;
[0031]
步骤3:配置crispr反应混合液:将0.4μl的浓度为1m的hepes缓冲液、0.18μl的浓度为1m的mgcl2溶液、1.6μl的10
×
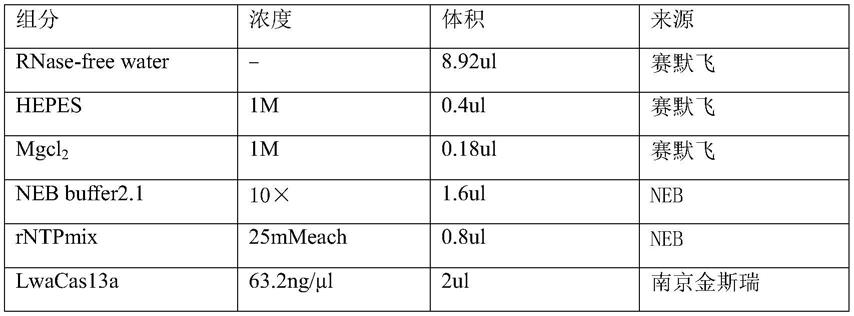
neb buffer2.1缓冲液、0.8μl的浓度为25um每个的rntp mix、2μl的浓度为的63.2ng/μl的lwacas13a、1μl的浓度为10ng/μl的cas13
‑
crrna、1μl的浓度为1um的lbacas12a、1μl的浓度为15ng/μl的cas12
‑
crrna,1μl的浓度为40u/μl的rnase inhibitor、0.1μl的浓度为50u/μl的t7 rna polymerase、0.1μl的浓度为100um的dna荧光探针、0.1μl的浓度为100um的rna荧光探针和8.92μl的无rna酶水混合;
[0032]
cas12a
‑
n
‑
crrna:
[0033]5’‑
uaauuucuacuaaguguagauauggcaccuguguaggucaa
‑3’
;
[0034]
cas13a
‑
s
‑
crrna:
[0035]5’‑
gauuuagacuaccccaaaaacgaaggggacuaaaaccauaagucacaugcaagaagacuacacc;
[0036]
rna荧光探针:
[0037]5’‑
fam
‑
mararurgrgrcmamararurgrgrcma
‑
bhq1
‑3’
;
[0038]
其中,m代表2位氧甲基修饰,r代表核糖核苷酸;
[0039]
dna荧光探针:
[0040]5’‑
vic
‑
ttattatt
‑
bhq1
‑3’
;
[0041]
步骤4:取2μl步骤2得到的核酸扩增产物加入到步骤3配置得到的crispr反应混合液,37℃孵育30分钟;通过下列方法进行判断:
[0042]
1、通过荧光定量pcr仪孵育并同时检测荧光,或直接水浴最后肉眼观察荧光变化;
[0043]
2、在crispr反应孵育开始时使用qpcr仪器读取fam通道下的荧光值,37℃孵育20个循环,每个循环间隔2分钟,每次循环结束记录一次荧光信号,通过最终荧光信号强度判断双靶点检测结果,即荧光值大于3000代表对应位点检测阳性,如fam通道荧光值大于3000,即cas13a所对应的新冠病毒s位点检测阳性,vic通道荧光值大于3000,即cas12a所对应的新冠病毒n位点检测阳性,荧光值小于2000代表对应位点检测阴性,若荧光值处于2000
‑
3000之间则重新检测一次,若荧光值依然为2000
‑
3000则判定对应位点检测阳性。
[0044]
优选的技术方案为:buffer a溶液配置方法为:向1l水中加入50mmol的tris缓冲液,100nmol的醋酸钾,20g的聚乙二醇粉末和2mmol二硫苏糖醇。
[0045]
由于上述技术方案运用,本发明与现有技术相比具有的优点是:
[0046]
本发明克服了detectr与sherlock系统的不兼容性,在不降低检测方法灵敏度与精确度的前提下,成功将源自瓦氏细单胞菌的lwacas13a(leptotrichia wadei cas13a)与源自链霉菌nd2006的lbacas12a(lachnospiraceae bacterium nd2006 cas12a)统一起来,实现了利用crispr系统对两个靶标位点进行同时检测。
附图说明
[0047]
图1 crispr双靶标检测方法的流程以及原理。
[0048]
图2 crispr检测位点的设计。
[0049]
图3交叉反应验证。
[0050]
图4 crispr双靶点检测方法的反应体系优化。
[0051]
图5金属离子对反应灵敏度及反应稳定性的优化。
[0052]
图6双靶点检测的灵敏度验证。
[0053]
图7实际样本验证。
具体实施方式
[0054]
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
[0055]
请参阅图1
‑
7。须知,本说明书所附图式所绘示的结构、比例、大小等,均仅用以配合说明书所揭示的内容,以供熟悉此技术的人士了解与阅读,并非用以限定本发明可实施的限定条件,故不具技术上的实质意义,任何结构的修饰、比例关系的改变或大小的调整。提供以下实施例以便更好地理解本发明,而非限制本发明。以下实施例中的实验方法如无特殊说明,均为常规方法。下述实施例中所用的实验材料如无特殊说明,均为常规生化试剂商店购买所得。
[0056]
实施例1:一种基于crispr/cas系统的新型冠状病毒双靶标快速检测方法及试剂盒
[0057]
试剂盒包括:
[0058]
cas13
‑
s
‑
for:5
’‑
gaaattaatacgactcactataggggctatcatcttatgtccttccctcagtcag
‑3’
;
[0059]
cas13
‑
s
‑
rev:5
’‑
aatggcaggagcagttgtgaagttcttttc
‑3’
;
[0060]
cas12
‑
n
‑
for:5
’‑
aatgtcgcgcattggcatggaagtcaca
‑3’
;
[0061]
cas12
‑
n
‑
rev:5
’‑
gacttgatctttgaaatttggatctttg
‑3’
;
[0062]
cas12a
‑
n
‑
crrna:
[0063]5’‑
uaauuucuacuaaguguagauauggcaccuguguaggucaa
‑3’
;
[0064]
cas13a
‑
s
‑
crrna:
[0065]5’‑
gauuuagacuaccccaaaaacgaaggggacuaaaaccauaagucacaugcaagaagacuacacc;
[0066]
rna荧光探针:5
’‑
fam
‑
mararurgrgrcmamararurgrgrcma
‑
bhq1
‑3’
;
[0067]
其中,m为2位氧甲基修饰,r为rna;
[0068]
dna荧光探针:5
’‑
vic
‑
ttattatt
‑
bhq1
‑3’
;
[0069]
buffer a溶液、bufferb溶液、hepes缓冲液、mgcl2溶液、10
×
neb buffer2.1缓冲
液、rnase inhibitor、t7 rna polymerase和无rna酶水。
[0070]
优选的技术方案为:buffer a溶液配置方法为:向1l水中加入50mmol的tris缓冲液,100nmol的醋酸钾,20g的聚乙二醇粉末和2mmol二硫苏糖醇;bufferb溶液为浓度为280mm的醋酸镁溶液。
[0071]
如图1所示,展示了基于crispr的双靶标检测的流程及原理。首先提取待检样本的核酸,然后通过rt
‑
raa对提取的核酸进行双位点(a/b位点)的恒温扩增以放大待检测信号,获得的恒温扩增产物被加入已配置好的crispr检测体系,lbacas12a与lwacas13a在各自的crrna的引导下识别各自的靶向核酸并激活对应的非特异性切割能力。crrna是一段引导rna,由固定的骨架(scaffold)以及一段与靶向序列互补的区域(spacer)构成。如图所示,lbacas12a通过cas12acrrna识别a位点并激活非特异性切割能力切割周围的单链dna探针产生荧光信号(vic)报告a位点的检测结果。lwacas13a通过cas13acrrna识别b位点并激活非特异性切割能力切割周围的单链rna探针产生荧光信号(fam)报告b位点的检测结果。双靶点的检测可在同一管中同时进行,通过荧光收集仪器进行采集。
[0072]
一种基于crispr/cas系统的新型冠状病毒双靶标快速检测方法,包括以下技术步骤。
[0073]
(1)提取新冠病毒核酸
[0074]
采集的样本保存于采集管中,将拭子头浸入含2
‑
3ml病毒保存液中(也可使用等渗盐溶液),尾部弃去,旋紧管盖。标本应尽快进行核酸提取并检测,可在24小时内检测的标本可置于4℃保存;24小时内无法检测的标本则应置于
‑
70℃或以下保存。使用rna提取试剂盒进行核酸提取,以试剂盒mini kit(qiagen,cat no.74106)为例:取200μl病毒保存液加入350μlbufferrlt吹打混匀,再加入550μl70%浓度的无水乙醇沉淀病毒rna。获得的浑浊悬液过滤柱离心,12000rpm,2min,4℃。先后使用bufferrw1与bufferrpe洗脱杂质,最后用80μl不含rna酶的水加至吸附柱中,通过离心溶解洗脱病毒核酸。
[0075]
(2)rt
‑
raa引物设计
[0076]
rt
‑
raa(reverse
‑
transcription
‑
recombinase
‑
aid amplification)即逆转录
‑
重组酶介导的扩增反应。即逆转录酶将rna逆转录成cdna,然后在多个重组酶与特异性的rt
‑
raa引物的介导下对目的片段进行37℃恒温扩增。rt
‑
raa反应是crispr检测方法中的第一步,起到放大信号从而提高检测灵敏度的作用。
[0077]
rt
‑
raa引物的设计遵循以下原则:1.引物长30
‑
35个碱基;2.引物gc含量为30%<gc<70%;3.扩增产物长度范围在100bp
‑
200bp之间;4.扩增区域需要gc含量为40%<gc<60%,避免单一重复序列以及回文序列。围绕检测位点设计上下游引物各一组。
[0078]
本发明由于是双靶点核酸检测,所以需要设计两组rt
‑
raa扩增引物,一组为cas12a靶向的n基因的rt
‑
raa引物,一组为cas13a靶向的s基因的rt
‑
raa引物。由于cas13a识别的是rna,还需要将恒温扩增得到的dna片段转录为rna,所以cas13a的rt
‑
raa引物在上游引物的5’端增加了一段t7启动子识别位点(大写字母表示)用于crispr检测时进行rna转录。
[0079]
cas13
‑
s
‑
for:5
’‑
gaaattaatacgactcactataggggctatcatcttatgtccttccctcagtcag
‑3’
;
[0080]
cas13
‑
s
‑
rev:5
’‑
aatggcaggagcagttgtgaagttcttttc
‑3’
;
[0081]
cas12
‑
n
‑
for:5
’‑
aatgtcgcgcattggcatggaagtcaca
‑3’
;
[0082]
cas12
‑
n
‑
rev:5
’‑
gacttgatctttgaaatttggatctttg
‑3’
。
[0083]
(3)rt
‑
raa双靶点核酸扩增
[0084]
使用杭州众测rt
‑
raa核酸扩增基础试剂盒以及对应的rt
‑
raa引物对提取的样本核酸进行扩增,37℃孵育20分钟。具体操作步骤如下:
[0085]
50μl rt
‑
raa扩增体系,取装有蛋白酶冻干粉的反应管,向其中依次加入37.5μl buffer a溶液(50mm tris ph 7.9,100nm醋酸钾(potassium acetate),5%聚乙二醇(peg),2mm二硫苏糖醇(dtt)),两种靶位点rt
‑
raa上游引物各2μl(10μm),两种靶位点rt
‑
raa下游引物各2μl(10μm)以及2μl待检核酸。加2.5μl bufferb溶液(280mm醋酸镁溶液(magnesiumacetate))至管盖上,盖上管盖后将反应管瞬离即可启动扩增反应。将反应管39℃水浴孵育20分钟或直接握在手心利用体温孵育20分钟。
[0086]
(4)双靶点检测方法crispr位点的设计
[0087]
设计用于检测的crrna:lbacas12a的检测需要在待识别序列上有一个用于识别的pam区域,pam区域的序列为tttn。lwacas13a的crrna设计无特殊要求。因此通过将新冠病毒基因组序列(nc_045512)与其它几种冠状病毒比对分析包括hcov
‑
229e(nc_002645),hcov
‑
hku1(nc_006577),hcov
‑
nl63(nc_005831),hcov
‑
oc43(nc_006213),sars
‑
cov(nc_004718)and bat sars
‑
like coronavirus(mg772933),避开同源序列最终设计出针对lbacas12a以及lwacas13a的检测位点。
[0088]
本发明最终设计的检测位点如下:
[0089]
cas12a
‑
n
‑
crrna:
[0090]5’‑
uaauuucuacuaaguguagauauggcaccuguguaggucaa
‑3’
;
[0091]
cas13a
‑
s
‑
crrna:
[0092]5’‑
gauuuagacuaccccaaaaacgaaggggacuaaaaccauaagucacaugcaagaagacuacacc
‑3’
。
[0093]
(5)配置crispr反应体系用于crispr双靶点快速检测
[0094]
由于cas12a体系与cas13a体系对反应体系不同,达到最佳灵敏度时需要不同的离子及离子浓度,在保证检测方法灵敏度与稳定性的情况下,本发明通过尝试不同离子的加入最终成功将lbacas12a与lwacas13a整合并可用于新冠病毒的快速超敏双靶标检测。
[0095]
按如下表格所述成分,体积以及浓度配置crispr双靶标检测体系:
[0096]
crispr反应混合液配置体系如下:
[0097]
[0098][0099]
rna荧光探针(fam
‑
rna探针):(m为2位氧甲基修饰,r为rna);
[0100]5’‑
fam
‑
mararurgrgrcmamararurgrgrcma
‑
bhq1
‑3’
;
[0101]
dna荧光探针(vic
‑
dna探针):
[0102]5’‑
vic
‑
ttattatt
‑
bhq1
‑3’
。
[0103]
取18ul检测体系,向其中加入2ul体积rt
‑
raa扩增产物,37℃孵育30分钟,通过fam,vic通道进行荧光监测,得到监测结果。
[0104]
(6)crispr双靶点的荧光检测及结果读取
[0105]
取2μl核酸扩增产物加入到配制好的18μlcrispr反应混合液,37℃孵育30分钟。可在crispr反应孵育开始时使用qpcr仪器(伯乐cfx96)读取fam与vic通道下的荧光值,37℃孵育20个循环,每个循环间隔2.5分钟,每次循环结束记录一次荧光信号。通过最终荧光信号强度判断双靶点检测结果,
[0106]
即荧光值大于3000代表对应位点检测阳性。如fam通道荧光值大于3000,即cas13a所对应的新冠病毒s位点检测阳性。vic通道荧光值大于3000,即cas12a所对应的新冠病毒n位点检测阳性。荧光值小于2000代表对应位点检测阴性,若荧光值处于2000
‑
3000之间则重新检测一次,若荧光值依然为2000
‑
3000则判定对应位点检测阳性。所述的fam通道,是qpcr仪器读取荧光时的一种荧光检测通道,读取波长为450nm
‑
490nm的荧光信号。vic通道读取波长为500
‑
535nm。
[0107]
crispr双靶点快速检测方法的建立
[0108]
1.材料
[0109]
rt
‑
raa扩增引物,crrna及单链探针由南京擎科生物公司以及南京金斯瑞生物公司合成。rt
‑
raa核酸基础扩增试剂盒购自杭州众测生物公司。lbacas12a蛋白购自于neb。lwacas13a蛋白购自于南京金斯瑞生物公司。
[0110]
2.方法与结果
[0111]
2.1:rt
‑
raa引物的设计
[0112]
根据genbank中已经公布的新冠病毒原始毒株(nc_045512.2)序列,设计s位点以及n位点的rt
‑
raa扩增引物,按照rt
‑
raa的要求设计每个靶位点对应的上游引物与下游引物,具体序列如下表:
[0113][0114]
2.2 lbacas12acrrna的设计以及lwacas13acrrna的设计
[0115]
为了保证设计的特异性,避免同种类病毒的同源片段导致的假阳性问题,在设计crrna时,我们将新型冠状病毒基因组序列(nc_045512)与其它几种冠状病毒进行比对分析,包括hcov
‑
229e(nc_002645),hcov
‑
hku1(nc_006577),hcov
‑
nl63(nc_005831),hcov
‑
oc43(nc_006213),sars
‑
cov(nc_004718)和batsars
‑
like coronavirus(mg772933),从中选出新型冠状病毒特有的序列作为crrna位点。另外,在位点设计时,lbacas12a的检测需要在待识别序列上有一个用于识别的pam区域,pam区域的序列为tttn。lwacas13a的crrna设计则无特殊要求。
[0116]
本发明最终设计的crrna检测位点如下:
[0117]
cas12a
‑
n
‑
crrna:
[0118]5’‑
uaauuucuacuaaguguagauauggcaccuguguaggucaa
‑3’
[0119]
cas13a
‑
s
‑
crrna:
[0120]5’‑
gauuuagacuaccccaaaaacgaaggggacuaaaaccauaagucacaugcaagaagacuacacc
[0121]
其与多种冠状病毒的序列比对如图2所示
[0122]
2.3实验验证lbacas12a与lwacas13a的非特异性切割不会发生交叉反应
[0123]
由于lbacas12a激活后仅切割周围的单链dna,lwacas13a激活后仅切割周围的单链rna,理论上不会发生交叉反应导致lbacas12a切割单链rna,lwacas13a切割单链dna。这是将lbacas12a与lwacas13a整合为同一系统并进行双靶点检测的理论前提。所以首先通过实验验证lbacas12a与lwacas13a激活后不会发生对不同探针底物的交叉反应。在完整的detectr实验中,加入rna探针(fam标记);在完整的sherlock实验中加入dna探针(vic标记),看是否会发生对底物的交叉切割。
[0124]
2.4不同buffer体系的尝试
[0125]
将lbacas12a与lwacas13a整合进同一体系首先要对反应体系的离子成分进行摸索,得到一个既能兼顾lbacas12a又能兼顾lwacas13a的反应体系。本发明在兼顾lwacas13a反应所需的几种成分(hepes,rntpmix,t7 rna聚合酶和rna酶抑制剂)的基础上,按不同比例掺入lbacas12a的反应体系的不同成分如nacl,tris
‑
hcl,mgcl2,bsa等。
[0126]
使用10000cp/ul的病毒基因组进行体系兼容性验证。病毒基因组首先通过rt
‑
raa恒温扩增,然后加入到配制好的多组crispr双靶标反应体系,通过对fam/vic通道的荧光信号监测判断最佳的反应体系。即在固定病毒浓度下,监测结果荧光值最高,监测结果最稳定的组合。由实验得知,在引入80%的lbacas12a反应体系时,双靶点检测的效率以及稳定性最好。两个位点均能有效被检测到。如图4所示
[0127]
2.5不同二价金属离子的体系优化
[0128]
金属离子是酶的辅基或激活剂,可以帮助稳定构象,构成酶的活性中心,亦或是起连接作用,作为桥梁将酶与底物整合连接起来。常见的酶促二价金属离子有mg,ca,mn,ni,zn,co,cu等。本发明对mg,ca,mn,ni等二价离子进行筛选并最终得出最适宜crispr双靶点检测方法的金属离子及其浓度。本实施例中,在已摸索好的buffer体系下,通过设置单一变量,验证不同金属离子对双靶点crispr检测的影响,本实施例中共设置了5组变量,分别探究了1mmmg2+,10mmmg2+,1mmca2+,1mm mn2+,1mm ni2+的对检测灵敏度的影响。实验结果显示mg2+对反应灵敏度提升最大,高浓度mg2+比低浓度mg2+更能提高检测的灵敏度与稳定性。如图5所示。
[0129]
2.6双通道有效灵敏度验证
[0130]
确定了crispr双靶点检测方法的最佳反应体系成分后,使用体外转录的新冠病毒s片段以及n片段rna模板,梯度稀释至8个不同浓度梯度:s/n,0cp/ul,0.7/1cp/ul,7/10cp/ul,17.5/25cp/ul,35/50cp/ul,70/100cp/ul,700/1000cp/ul,7000/10000cp/ul。通过完整的crispr双靶点检测方法对不同浓度的稀释模板进行检测,验证检测方法的灵敏度。由图可见本发明中新冠crispr双靶点检测方法可稳定检出两个位点的灵敏度分别是:cas13a 7cp/ul,cas12a 25cp/ul。如图6所示
[0131]
2.7双通道实际样本验证
[0132]
实际临床样本验证:使用crispr双靶点检测方法对24个新冠临床样本进行检测。对采集到的24个新冠病毒临床样本进行核酸提取,使用本发明中提到的新冠病毒s位点与n位点的rt
‑
raa引物对2ul体积的样本核酸进行恒温扩增,扩增得到的rt
‑
raa产物取2ul体积加入配制好的18ulcrispr双靶点检测体系中,37℃孵育1小时。在孵育开始时,通过荧光读取机器读取fam与vic通道的荧光数值。使用crispr双靶点检测方法检测24例临床样本结果显示16例阳性,8例阴性,与qpcr结果一致。如图7所示
[0133]
图1:crispr双靶标检测方法的流程以及原理
[0134]
即先对样本进行处理以获得核酸,然后使用两对rt
‑
raa引物对检测的两个核酸位点同时进行恒温扩增,最后将获得的rt
‑
raa产物加入配制好的双靶点crispr反应体系,37℃孵育一小时并检测fam与vic通道的荧光信号,得出检测结果。
[0135]
图2:crispr检测位点的设计
[0136]
为了保证检测方法的特异性,避免因为序列相似导致的检测假阳性。本发明在设计crrna时,除了考虑sars
‑
cov
‑
2的序列特征以外,还将sars
‑
cov
‑
2的序列与多种其它冠状病毒进行同源比对,包括包括人类冠状病毒229e,hku1,nl63,oc43,sars
‑
cov以及蝙蝠类sars冠状病毒。如图蓝色部分为比对后的同源序列,红色部分为基因组间的差异序列。
[0137]
图3:交叉反应验证:
[0138]
证明了双通道检测方法中的两个关键crispr蛋白酶lwacas13a以及lbacas12a在激活后不会对各自的反应底物发生交叉切割,从而保证了crispr双靶点检测的理论可行性。
[0139]
图4:crispr双靶点检测方法的反应体系优化
[0140]
在本实施例中,在cas13a蛋白的反应体系上,引入不同比例的cas12a蛋白反应体系,分别是100%,80%,60%,40%以及20%。使用固定浓度的新冠病毒模板(10000cp/ul),
先经过双位点的rt
‑
raa恒温扩增,再取2ul体积的rt
‑
raa扩增产物加入到不同成分比例的crispr双靶点检测体系中,37℃孵育1小时,孵育全程每隔2分钟检测一次fam与vic通道的荧光值。其中lwacas13a切割fam标记的rna探针,lbacas12a切割vic标记的dna探针,所以fam阳性即代表lwacas13a检测的基因位点阳性,vic阳性即代表lbacas12a检测的基因位点阳性。
[0141]
图5:金属离子对反应灵敏度及反应稳定性的优化
[0142]
验证mg,ca,mn,ni等不同二价离子对crispr双靶点检测灵敏度的提升效果
[0143]
图6:双靶点检测的灵敏度验证
[0144]
确定crispr双靶点检测方法的最佳反应体系成分后,通过完整的crispr双靶点检测方法对不同浓度的稀释模板进行检测,验证检测方法的灵敏度。由图可见本发明中新冠crispr双靶点检测方法可稳定检出两个位点的灵敏度分别是:cas13a 7cp/ul,cas12a 25cp/ul。
[0145]
图7:实际样本验证
[0146]
用crispr双靶点检测方法同时检测24例新冠临床样本,共检出16例阳性与8例阴性样本,与荧光定量结果一致。
[0147]
实施例2:一种基于crispr/cas系统的新型冠状病毒双靶标快速检测方法及试剂盒
[0148]
一种基于crispr/cas系统的新型冠状病毒双靶标快速检测的试剂盒,所述试剂盒包括:
[0149]
两组rt
‑
raa扩增引物:
[0150]
cas13
‑
s
‑
for:5
’‑
gaaattaatacgactcactataggggctatcatcttatgtccttccctcagtcag
‑3’
;
[0151]
cas13
‑
s
‑
rev:5
’‑
aatggcaggagcagttgtgaagttcttttc
‑3’
;
[0152]
cas12
‑
n
‑
for:5
’‑
aatgtcgcgcattggcatggaagtcaca
‑3’
;
[0153]
cas12
‑
n
‑
rev:5
’‑
gacttgatctttgaaatttggatctttg
‑3’
;
[0154]
两种crispr特异性检测的crrna:
[0155]
cas12a
‑
n
‑
crrna:
[0156]5’‑
uaauuucuacuaaguguagauauggcaccuguguaggucaa
‑3’
;
[0157]
cas13a
‑
s
‑
crrna:
[0158]5’‑
gauuuagacuaccccaaaaacgaaggggacuaaaaccauaagucacaugcaagaagacuacacc
‑3’
;
[0159]
rna荧光探针:5
’‑
fam
‑
mararurgrgrcmamararurgrgrcma
‑
bhq1
‑3’
;
[0160]
其中,m代表2位氧甲基修饰,r代表核糖核苷酸;
[0161]
dna荧光探针:5
’‑
vic
‑
ttattatt
‑
bhq1
‑3’
。
[0162]
还包括buffer a溶液和bufferb溶液;buffer a溶液配置方法为:向1l水中加入50mmol的tris缓冲液,100nmol的醋酸钾,20g的聚乙二醇粉末和2mmol二硫苏糖醇;bufferb溶液为浓度为280mm的醋酸镁溶液。
[0163]
还包括:hepes缓冲液、mgcl2溶液、10
×
neb buffer2.1缓冲液、rnase inhibitor、t7rna polymerase、无rna酶水。
[0164]
一种基于crispr/cas系统的新型冠状病毒双靶标快速检测方法,包括下列步骤:
[0165]
步骤1:将待检测样本浸入病毒保存液中,然后用rna提取试剂盒进行核酸提取,得到待检核酸;待检测样本由门把手上提取得到。
[0166]
步骤2:向一容纳有蛋白酶冻干粉的反应管中,加入37.5μl的buffer a溶液,2μl的cas13
‑
s
‑
for,2μl的cas13
‑
s
‑
rev,2μl的cas12
‑
n
‑
for,2μl的cas12
‑
n
‑
rev以及2μl待检核酸,然后加2.5μl的bufferb溶液,盖上反应管的管盖后进行扩增反应,得到核酸扩增产物;
[0167]
cas13
‑
s
‑
for:5
’‑
gaaattaatacgactcactataggggctatcatcttatgtccttccctcagtcag
‑3’
;
[0168]
cas13
‑
s
‑
rev:5
’‑
aatggcaggagcagttgtgaagttcttttc
‑3’
;
[0169]
cas12
‑
n
‑
for:5
’‑
aatgtcgcgcattggcatggaagtcaca
‑3’
;
[0170]
cas12
‑
n
‑
rev:5
’‑
gacttgatctttgaaatttggatctttg
‑3’
;
[0171]
步骤3:配置crispr反应混合液:将0.4μl的浓度为1m的hepes缓冲液、0.18μl的浓度为1m的mgcl2溶液、1.6μl的10
×
neb buffer2.1缓冲液、0.8μl的浓度为25um每个的rntp mix、2μl的浓度为的63.2ng/μl的lwacas13a、1μl的浓度为10ng/μl的cas13
‑
crrna、1μl的浓度为1um的lbacas12a、1μl的浓度为15ng/μl的cas12
‑
crrna,1μl的浓度为40u/μl的rnase inhibitor、0.1μl的浓度为50u/μl的t7 rna polymerase、0.1μl的浓度为100um的dna荧光探针、0.1μl的浓度为100um的rna荧光探针和8.92μl的无rna酶水混合;
[0172]
cas12a
‑
n
‑
crrna:
[0173]5’‑
uaauuucuacuaaguguagauauggcaccuguguaggucaa
‑3’
;
[0174]
cas13a
‑
s
‑
crrna:
[0175]5’‑
gauuuagacuaccccaaaaacgaaggggacuaaaaccauaagucacaugcaagaagacuacacc;
[0176]
rna荧光探针:
[0177]5’‑
fam
‑
mararurgrgrcmamararurgrgrcma
‑
bhq1
‑3’
;
[0178]
其中,m代表2位氧甲基修饰,r代表核糖核苷酸;
[0179]
dna荧光探针:
[0180]5’‑
vic
‑
ttattatt
‑
bhq1
‑3’
;
[0181]
步骤4:取2μl步骤2得到的核酸扩增产物加入到步骤3配置得到的crispr反应混合液,37℃孵育30分钟;通过下列方法进行判断:
[0182]
1、通过荧光定量pcr仪孵育并同时检测荧光,或直接水浴最后肉眼观察荧光变化;
[0183]
2、在crispr反应孵育开始时使用qpcr仪器读取fam通道下的荧光值,37℃孵育20个循环,每个循环间隔2分钟,每次循环结束记录一次荧光信号,通过最终荧光信号强度判断双靶点检测结果,即荧光值大于3000代表对应位点检测阳性,如fam通道荧光值大于3000,即cas13a所对应的新冠病毒s位点检测阳性,vic通道荧光值大于3000,即cas12a所对应的新冠病毒n位点检测阳性,荧光值小于2000代表对应位点检测阴性,若荧光值处于2000
‑
3000之间则重新检测一次,若荧光值依然为2000
‑
3000则判定对应位点检测阳性。
[0184]
以上所述者仅为用以解释本发明之较佳实施例,并非企图具以对本发明做任何形式上之限制,是以,凡有在相同之发明精神下所作有关本发明之任何修饰或变更,皆仍应包括在本发明意图保护之范畴。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1