无水相制备氯乙醛及氯乙醛缩醇、2,5-二羟基-1,4-二噻烷和2-氨基噻唑的方法与流程
1.本技术涉及药物中间体合成领域,更具体地说,它涉及无水相制备氯乙醛及氯乙醛缩醇、2,5-二羟基-1,4-二噻烷和2-氨基噻唑的方法。
背景技术:
2.氯乙醛是一种重要的药物中间体,在药物制备中具有重要的意义。市售氯乙醛多为水溶液或混合溶液,其在反应过程中,三废产生总量巨大,且当反应需要无水体系时,无法满足反应需求。
3.截止到目前,研究人员在制备无水氯乙醛方面进行了研究,主要通过三聚氯乙醛在高温下催化裂解的方式进行,该方法需要的催化温度一般较高,超过氯乙醛的沸点(85℃),因此其在制备过程中,生成的氯乙醛容易发生聚合,为减少聚合的发生,往往需要大量的溶剂对氯乙醛进行稀释,降低其聚合,如此一来,产生的三废总量又较大,不利于工业大规模生产。
技术实现要素:
4.为了得到一种适用于工业大规模生产的无水相氯乙醛,本技术提供无水相制备氯乙醛及氯乙醛缩醇、2,5-二羟基-1,4-二噻烷和2-氨基噻唑的方法。
5.首先,本技术提供了无水相制备氯乙醛的制备方法,通过将含有1,2-二氯乙醇醋酸酯与裂解反应催化剂加热使之进行热裂解反应,加热至52~80℃,使1,2-二氯乙醇醋酸酯裂解为乙酰氯和氯乙醛,具体反应如式ⅰ所示;所述裂解反应催化剂为路易斯酸类催化剂。
6.在上述技术方案中,通过1,2-二氯乙醇醋酸酯催化裂解的方式,从而制备得到氯乙醛,在该反应中,裂解反应在较低的温度下进行,整体反应温度低于氯乙醛的沸点,因此反应过程中,氯乙醛不易重新发生聚合,因此可以在少溶剂甚至无溶剂的体系中,得到较高的氯乙醛产率。
7.由于反应整体温度降低,且溶剂用量减少,因此在上述技术方案中,最终产生的三废较少,得到的副产物主要为乙酰氯,同样是工业中需要运用的产品。反应收率较高,副产物少,且乙酰氯和氯乙醛沸点差别较大,通过蒸馏即可分离,得到两个组分均具有较好的纯度,有助于降低企业的生产成本。
8.可选的,所述裂解反应催化剂为发烟硫酸、磷酸、对甲苯磺酸、甲磺酸、三氟甲磺酸、冰醋酸、甲酸、三氯化铝、四氯化锡、氯化铁、氯化锌、溴化锌、蒙脱石k10中的至少一种,或发烟硫酸、浓磷酸、对甲苯磺酸、甲磺酸、三氟甲磺酸、冰醋酸、甲酸、三氯化铝、四氯化锡、
氯化铁、氯化锌、溴化锌、蒙脱石k10中任意数种形成的混合体系;所述裂解反应催化剂的用量为1,2-二氯乙醇醋酸酯质量的0.1~10%。
9.选取上述裂解反应催化剂,在催化裂解步骤中,有助于提高氯乙醛的产率,且氯乙醛最终可以通过分馏的方式,从上述成分中分离,有助于提高最终产品的纯度。
10.可选的,在热裂解反应过程中,反应温度为53~55℃。
11.生成的副产物乙酰氯的上述反应温度低于上述技术方案的反应温度,使得反应进行过程中,乙酰氯可以被不断地蒸出,从而使反应平衡不断向裂解的方向,进一步提高反应的产率。
12.可选的,热裂解反应过程中,1,2-二氯乙醇醋酸酯和热裂解催化剂分散于非水溶剂中,非水溶剂为二氯甲烷、三氯甲烷或四氯化碳中的至少一种,1,2-二氯乙醇醋酸酯在所述非水溶剂中的浓度为0.1~20m。
13.在上述技术方案中,在适量溶剂中进行如式ⅰ所示的反应,反应产率更高,且由于反应组分与非水溶剂的沸点相差较大,反应过程中使用的溶剂可以在后续过程中回收并重新利用,有助于企业节约成本。
14.可选的,在热裂解反应过程中,不额外添加溶剂,直接以1,2-二氯乙醇醋酸酯为体系进行反应。
15.在上述技术方案中,没有添加溶剂,直接在1,2-二氯乙醇醋酸酯的体系中进行反应,反应可以顺利进行的同时,由于没有溶剂的参与,后续提纯较为简单,产生的废液较少,且容易分离。
16.可选的,1,2-二氯乙醇醋酸酯通过如下方法制备:将醋酸乙烯酯在相转移催化剂作用下,与-40~20℃下,与氯气进行加成反应,得到1,2-二氯乙醇醋酸酯,具体反应如式ⅱ所示;所述相转移催化剂为季铵盐催化剂或季磷盐催化剂。
17.在上述技术方案中,通过加成反应制备得到1,2-二氯乙醇醋酸酯,反应过程较为简单,且反应温度容易控制,整体产率较高,且在有无溶剂的条件下均可以直接进行反应,工业适用性好。
18.可选的,1,2-二氯乙醇醋酸的制备步骤中,还加入非水溶剂,所述非水溶剂为二氯甲烷、三氯甲烷或四氯化碳中的至少一种,醋酸乙烯酯在所述非水溶剂中的浓度为0.1~10m。
19.在上述技术方案中,通过加入一定量的非水溶剂,有助于提高加成反应的产率。
20.可选的,所述相转移催化剂为四丁基溴化铵或四丁基氯化铵,所述相转移催化剂的用量为醋酸乙烯酯质量的0.1~5%。
21.在-15~15℃范围内进行处理,整体副反应较少,产物较纯且具有较好的产率。温度容易控制,能耗较低。
22.可选的,加成反应的反应温度为-15~15℃。
23.四丁基溴化铵和四丁基氯化铵价格较为便宜,且采用这两种相转移催化剂有助于
提高1,2-二氯乙醇醋酸的产率。
24.另外,本技术还涉及无水相制备氯乙醛衍生产品的制备方法,先按照上述方法,制备得到氯乙醛,再通过氯乙醛制备氯乙醛缩醇、2,5-二羟基-1,4-二噻烷、2-氨基噻唑盐酸盐中的任意一者。
25.第二,本技术提供一种制备氯乙醛缩醇的制备方法,采用如下方案:先按照上述方案制备得到氯乙醛,再将氯乙醛通入含有氯化氢的醇ⅰ溶液中,制备得到氯乙醛缩醇,所述醇ⅰ为一元伯醇或二元伯醇。
26.在上述技术方案中,采用无水相方法制备得到的氯乙醛中不含税,产生的废料较少,便于分离,减少了能耗并简化了分离步骤,具有较好的工业运用前景。
27.第三,本技术提供2,5-二羟基-1,4-二噻烷的制备方法,采用如下技术方案:先按照如权利要求1~9中任意一项所述的方法,制备得到氯乙醛,再在0~5℃下,将氯乙醛滴加到硫氢化钠的水溶液中,充分反应后制备得到2,5-二羟基-1,4-二噻烷。
28.在上述技术方案中,采用无水相制备得到的氯乙醛,氯乙醛无需进行前处理,且产生的副产物较少,产生的非水也较少,后续处理较为方便,具有较好的工业运用前景。
29.第四,本技术提供2,5-二羟基-1,4-二噻烷的制备方法,采用如下技术方案:先按照如权利要求1~9中任意一项所述的方法,制备得到氯乙醛,再在60~70℃下,将氯乙醛和硫脲的水溶液混合,制备得到2-氨基噻唑。
30.在上述技术方案中,采用无水相方法制备得到的氯乙醛,得到的最终产物纯度较好,收率较好,产生的副产物较少,具有良好的工业运用前景。
31.综上所述,本技术至少包括如下一种有益效果:1.在本技术中,采用先氯化,再加热裂解的方式,整个反应温度控制在80℃以下,减少了生成的氯乙醛再高温状态下的聚合,提高反应的产率,同时减少了溶剂大量使用的必要性,使得企业的生产成本大幅降低。
32.2.在本技术进一步设置中,通过设置步骤s2的反应温度为53~55℃,使之略高于乙酰氯的沸点,使反应过程中乙酰氯被不断地蒸出,从而使反应平衡不断向生成氯乙醛的方向前进,进一步提高了反应的产率。
33.3.在本技术的进一步设置中,通过本技术制得的氯乙醛,可以进一步合成氯乙醛缩醇、2,5-二羟基-1,4-二噻烷、2-氨基噻唑盐酸盐等化合物,具有较好的工业运用前景。
具体实施方式
34.以下结合和实施例对本技术作进一步详细说明。
35.实施例1,无水相制备氯乙醛的制备方法,具体包括如下步骤:s1、加成反应,将醋酸乙烯酯在相转移催化剂作用下,与氯气进行加成反应,得到1,2-二氯乙醇醋酸酯,具体反应如式ⅰ所示。
36.步骤s1具体如下:在0℃下,在三口瓶中加入醋酸乙烯酯860g(10mol),四丁基溴化铵8.6g,并加入10l二氯甲烷将其溶解,保持搅拌并在避光状态下通入10ml氯气(710g),在
整个反应过程中,控制反应温度低于15℃,反应2h后,完成反应,得到含有1,2-二氯乙醇醋酸酯的二氯甲烷溶液。对上述含有1,2-二氯乙醇醋酸酯的二氯甲烷溶液进行精馏处理,除去氯气和二氯甲烷(二氯甲烷可以回用),得到1,2-二氯乙醇醋酸酯1500.8g,收率95.6%。
37.对于该步反应,一般反应时间为1~4h,由于非水溶剂的用量和温度可以在一定范围内进行调整,因此反应时间也可以进行调整。当非水溶剂用量增大或反应温度降低时,反应时间可以适当延长,当非水溶剂用量减少或反应温度升高时,反应时间可以适当缩短。具体的反应时间可以通过实时点板监测确定,也可直接肉眼观察,当反应体系变为无色时,即可停止反应。
38.s2、热裂解反应,将1,2-二氯乙醇醋酸酯与裂解反应催化剂一同加热并使1,2-二氯乙醇醋酸酯裂解为乙酰氯和氯乙醛,反应如式ⅱ所示。
39.该反应具体如下:取1,2-二氯乙醇醋酸酯157.0g(1mol)溶解于1200ml二氯甲烷中,加入2.0g发烟硫酸,搅拌加热至53
±
1℃,反应5h,在反应过程中不断蒸出二氯甲烷的乙酰氯溶液,最终得到氯乙醛66.1g,收率84.2%。同时,精馏分离二氯甲烷和乙酰氯,可以得到乙酰氯69.0g,并对二氯甲烷进行回用。
40.氯乙醛在氘代氯仿中进行核磁共振氢谱监测,数据如下:1h nmr:[9.62(s,ch),4.07ppm(s,ch2)]。
[0041]
在上述反应中,氯气的用量以醋酸乙烯酯物质的量的1~1.05倍为宜,由于氯气具有毒性,因此尽量少用氯气,在实际生产中具有较大的意义,有助于后续安全地除去残留氯气。
[0042]
与步骤s1类似的,在步骤s2中,反应时间同样与非水溶剂的用量和反应温度有关,可以通过点板监测确定。一般情况下,反应时间为4~6h。
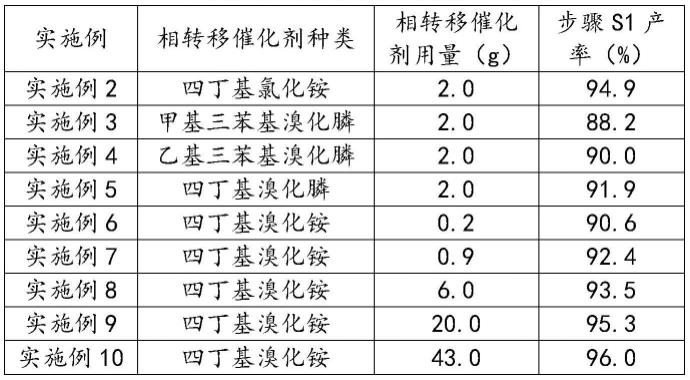
[0043]
实施例2~11,无水相制备氯乙醛的制备方法,与实施例1的区别在于,在步骤s1中,具体采用的相转移催化剂的种类和用量如表1所示。表1、调整相转移催化剂对反应的影响表1、调整相转移催化剂对反应的影响
[0044]
通过上述实验数据可知,四丁基氯化铵和四丁基溴化铵相较于甲基三苯基溴化膦和乙级三苯基溴化膦,具有更好的催化效果。其原因可能在于氮原子和磷原子的尺寸差别,导致季膦盐在参与反应过程中受到的位阻较大,且四丁基氯化铵和四丁基溴化铵价格较为便宜,是更好的工业选择。
[0045]
对于相转移催化剂的用量,通过实施例5~11和实施例1的比对可以看出,在相转移催化剂用量不断增加的情况下,反应的产率也有一定的提高,当相转移催化剂用量达到醋酸乙烯酯的5%时,继续增加相转移催化剂的用量,对于步骤s1的产率基本没有影响。
[0046]
实施例12,无水相制备氯乙醛的制备方法,与实施例1的区别在于,在步骤s1中,不额外添加非水溶剂,直接进行反应,反应完成后,无需精馏处理即得到1,2-二氯乙醇醋酸,产率为91.1%。
[0047]
实施例13,无水相制备氯乙醛的制备方法,与实施例12的区别在于,在步骤s1中,相转移催化剂替换为等质量的四丁基氯化铵。
[0048]
实施例14,无水相制备氯乙醛的制备方法,与实施例12的区别子啊,在步骤s1中,相转移催化剂替换为等质量的四丁基溴化膦。
[0049]
实施例15~23,无水相制备氯乙醛的制备方法,与实施例1的区别在于,在步骤s1中,非水溶剂的用量和种类如表2所示。表2、溶剂调整对步骤s1的影响
[0050]
通过上述实验数据可得,在步骤s1中,无论是否使用溶剂,都可以获得较高的产率,且其中大部分溶剂都可以回收再利用,有助于节约成本。使用溶剂相较于直接进行反应,可以获得略高的最终产率,此处应当是由于,在溶剂的作用下,可以使氯气更加充分地与醋酸乙烯酯进行反应,进而提高了反应产率。但是溶剂量的过大会导致反应变慢,且使用溶剂参与该步骤的反应,后续需要对溶剂进行提取,因此,在该步骤中具体是否使用溶剂,可以依照实际和企业是否具有相关设备需要确定,具有较好的灵活性。
[0051]
实施例24~28,无水相制备氯乙醛的制备方法,与实施例1的区别在于,对步骤s1中的反应温度进行了调整,具体如表3所示。表3、步骤s1温度调整对反应的影响
[0052]
结合上述实施例,并与实施例1进行对比,可以看到步骤s1中的温度在-15~15℃范围内具有较好的反应效果,产率较高的同时,反应时间也较快。在更大尺度内,当温度处于-40~20℃范围内,反应均具有较好的产率,但是当温度较低时,反应较慢,而温度较高时,产率则有所下降。当温度超过20℃时,产率会进一步大幅下降。
[0053]
进一步地,对步骤s2进行调整,得到如下实施例。
[0054]
实施例29~42,无水相制备氯乙醛的制备方法,与实施例1的区别在于,在步骤s2中,选用了不同的热裂解催化剂,具体如表4所示。
[0055]
实施例43~56,无水相制备氯乙醛的制备方法,与实施例29~42的区别在于,在步骤s2中,不额外添加非水溶剂,在反应过程中,不断蒸出乙酰氯,最终得到的产物即为氯乙醛,进一步精馏后可以得到较纯的氯乙醛。在实施例43~56之间采用了不同的热裂解催化剂,具体如表4所示。表4、实施例29~56中对非水溶剂和热裂解催化剂的调整结果
[0056]
通过实施例29~56之间进行对比,可以看到,在步骤s2中选用不同的热裂解催化剂,其中的效果有一定的差别,实际生产过程中可以依照催化剂的价格进行选取。由于发烟硫酸价格较低,后续分离较为方便,因此在大部分实施例中,依旧是以发烟硫酸作为热裂解催化剂使用。在各种热裂解催化剂中,产率最高的是三氟甲磺酸,可能是由于三氟甲磺酸提供质子吸收电子的能力最强。
[0057]
在实施例43~56中,则换用了无溶剂的体系,无溶剂体系中反应产率有明显的降低,但是对于部分催化剂,例如三氟甲磺酸,依旧可以使反应产率达到90%以上,因此生产厂家可以依照自身具有的设备条件,涉及适合自身的合成路线,使得该方案具有较好的工业运用前景。
[0058]
进一步地,在实施例31、实施例32和实施例44和实施例45的基础上,调整催化剂的用量,得到如下实施例。
[0059]
实施例57~62,无水相制备氯乙醛的制备方法,与实施例29的区别在于,对热裂解催化剂的用量进行了调整,具体调整如表5所示。
[0060]
实施例63~68,无水相制备氯乙醛的制备方法,与实施例30的区别在于,对热裂解催化剂的用量进行了调整,具体调整如表5所示。
[0061]
实施例69~74,无水相制备氯乙醛的制备方法,与实施例44的区别在于,对热裂解催化剂的用量进行了调整,具体调整如表5所示。
[0062]
实施例75~80,无水相制备氯乙醛的制备方法,与实施例45的区别在于,对热裂解催化剂的用量进行了调整,具体调整如表5所示。
表5、催化剂用量对步骤s2产率的影响
[0063]
通过上述实验数据可知,当热裂解催化剂用量在1,2-二氯乙醇醋酸酯用量的0.1~10%范围内,提高催化剂的用量,具有提高反应产率的效果,超过1,2-二氯乙醇醋酸酯10%质量范围时,继续提高催化剂的用量,对于反应效果提升较小。
[0064]
进一步地,以实施例59、65、71、76为基础,进一步对反应温度进行调整,得到如下实施例。
[0065]
实施例81~84,无水相制备氯乙醛的制备方法,与实施例59的区别在于,步骤s2的温度进行了调整,具体如表6所示。
[0066]
实施例85~88,无水相制备氯乙醛的制备方法,与实施例65的区别在于,步骤s2的温度进行了调整,具体如表6所示。
[0067]
实施例89~92,无水相制备氯乙醛的制备方法,与实施例71的区别在于,步骤s2的温度进行了调整,具体如表6所示。
[0068]
实施例93~96,无水相制备氯乙醛的制备方法,与实施例76的区别在于,步骤s2的温度进行了调整,具体如表6所示。
表6、热裂解温度对步骤s2产率的影响
[0069]
通过上述实施例可知,在53~55℃范围内,反应具有更高的产率,进一步提高反应温度时,会导致得到的氯乙醛容易发生聚合,进而使得产率降低,该现象在无溶剂条件下更加明显。
[0070]
进一步地,以实施例82为基础,对步骤s2中所用的非水溶剂的用量进行调整,得到如下实施例。
[0071]
在上述实施例中,选取较佳的实施例,实施例86和实施例90,对步骤s1和s2进行扩大生产,得到如下实施例。
[0072]
实施例97,无水相制备氯乙醛的制备方法,具体包括如下步骤:s1、加成反应,预先向反应釜中加入200l二氯甲烷,并冷却至0℃,在三口瓶中加入醋酸乙烯酯17.2kg(200mol),四丁基溴化铵172g,保持搅拌并在避光状态下通入10ml氯气(710g),并保持反应体系在0
±
5℃范围内,反应3h后,精馏处理,二氯甲烷通过加热脱除氯气后保留回用,得到1,2-二氯乙醇醋酸酯29.1kg,收率92.7%。
[0073]
s2、热裂解反应,在反应釜中,预先加入20l二氯甲烷,并将3.14kg1,2-二氯乙醇醋酸酯分批加入,随后加入80g发烟硫酸,搅拌加热至55
±
1℃,反应5h,在反应过程中不断蒸出二氯甲烷的乙酰氯溶液,最终得到氯乙醛1.35kg,收率85.3%。
[0074]
实施例98,无水相制备氯乙醛的制备方法,与实施例97的区别在于,步骤s2具体如下:将3.14kg1,2-二氯乙醇醋酸酯加入反应釜中,搅拌并升温至55
±
1℃,分批加入80g发烟硫酸,保持温度反应5h,在反应过程中乙酰氯不断蒸出,最终留在反应釜中的即为氯乙醛,最终得到氯乙醛1.30kg,产率81.9%。
[0075]
通过上述两个实施例可知,采用本技术中的方式可以适用于大规模生产,具有较
好的工业运用前景。
[0076]
进一步,设置如下实施例,制备氯乙醛缩醇。
[0077]
实施例98,制备氯乙醛缩醇的制备方法,具体如下:取按照实施例97中方法制备得到的氯乙醛缩醇78.5g(1mol),在30min内滴加到含有质量分数为10%的氯化氢甲醇溶液中,甲醇的质量为106.8g(3mol),在滴加过程中,控制氯化氢-甲醇溶液的温度为40℃,滴加结束后升温至50℃,继续反应3~4h直至充分反应,反应结束后加入氢氧化钠调节ph至中性,蒸馏蒸出甲醇和水,并精馏得到氯乙醛缩二甲醇117.47g,产率为94.3%,该反应的方程式如式ⅲ所示,产物在氘代氯仿中的核磁共振氢谱数据为:1h nmr:[5.39ppm(s ch3)4.51ppm(t,ch),3.57ppm(d,ch2),3.41ppm(s ch3)]。
[0078]
值得注意的是,此处氢氧化钠换用氢氧化钾、碳酸氢钠均可,对反应产率无明显影响。
[0079]
在反应中,采用了过量的甲醇作为溶剂,也可用其他不参与反应的溶剂体系进行。氯乙醛的滴加时间一般不少于10min,否则氯乙醛之间容易发生自聚合反应。一般直接在甲醇体系中反应时,反应所需时间较短,在溶剂中反应所需的时间则较长。
[0080]
氯化氢的浓度不低于5%,采用催化量浓度即可进行,氯化氢浓度过低会导致反应时间延长且反应产率降低。一般在氯乙醛滴加到甲醇体系中时,温度为30~40℃,滴加完毕后升温至50~60℃。
[0081]
实施例99,制备氯乙醛缩醇的制备方法,与实施例98的区别在于,用等物质的量乙醇(153.6g,3mol)替换甲醇,得到氯乙醛缩二乙醇,产率91.7%,反应式如式ⅳ所示。最终产物在氘代氯仿中的核磁共振氢谱数据如下:1h nmr:[4.63ppm(s 1h)3.66ppm(dd,4h),3.50ppm(s,2h),1.24ppm(s 6h)]。
[0082]
实施例100,制备氯乙醛缩醇的制备方法,与实施例98的区别在于,用1.5mol乙二醇(103.5g)替换甲醇,得到氯乙醛缩乙二醇,产率93.9%,反应式如式
ⅴ
所示。制备得到的最终产物在氘代氯仿中的核磁共振氢谱数据如下:1h nmr:[5.16ppm(t1h),4.05ppm(d,2h),3.96ppm(d,2h),3.55ppm(d 2h)]。
[0083]
实施例101,制备氯乙醛缩醇的制备方法,与实施例98的区别在于,用3mol叔丁醇(200.1g)替换甲醇,得到氯乙醛缩二乙醇,产率89.1%反应式如式ⅵ所示。
[0084]
在上述实施例中,均采用无水相氯乙醛制备氯乙醛缩醇,具有较高的产率的同时后处理工艺较为简单,反应结束后直接蒸出多余的甲醇和水即可,产生的水较少,需要处理的废液量较少。另外,采用水相氯乙醛进行制备,得到对比例1如下。
[0085]
对比例1,氯乙醛缩二甲醇的制备方法,包括如下过程:(1)氯化:在2l三口瓶中加入乙酸乙烯酯860g、3-(3,5-二叔丁基-4-羟基苯基)丙酸8.6g,8.6g氯化苄基三苯基膦;避光并搅拌下,控15-20℃,通氯气710g,用时4小时。
[0086]
(2)醇解:将氯化液滴入1280g甲醇中,水浴控温不超过50℃;1小时滴毕,撤去水浴,在45-50℃继续搅拌2小时。
[0087]
(3)中和:将醇解液降温到8℃,冰水浴控温不超10℃,滴入850g20%的氨水直到反应液的ph值达到8;过滤生成的氯化铵,晾干得到氯化铵480g。
[0088]
(4)分液:滤液分出下层有机相,有机相用200g水+60g氯化铵的水溶液洗涤,静置分液,分出下层油相。
[0089]
(5)精馏:油相常压精馏,收集50-65℃馏分808g,主要是乙酸甲酯和约20%甲醇的混合物,蒸到釜底70℃;逐渐减压到0.09mpa,收集过度馏分80g(主要是甲醇、氯乙醛混合物);等压力恒定在0.09mpa,塔顶温度稳定在65℃,收集氯乙醛缩二甲醇1071g,收率86.0%。
[0090]
在对比例中,产生了乙酸甲酯和甲醇的混合物,该体系为共沸体系,不利于直接精馏分离套用,需要引入新的溶剂或水进行精馏,且其体系中,本身即含有大量的水,反正整体耗能较大,同时也增加了污染的风险。
[0091]
实施例102,制备2,5-二羟基-1,4-二噻烷的制备方法,具体如下:配置1180g含有2.1mol硫氢化钠的水溶液,通过冰水浴降温至0℃,取按照实施例97中的方法制备得到的氯乙醛157g(2mol),在2h内滴加到上述体系中,滴加过程中控制温度不高于5℃,滴加结束后,继续保持温度不高于5℃的反应温度,充分反应,2h后反应完全,蒸馏除去水,用甲醇精制得到白色固体即为2,5-二羟基-1,4-二噻烷,产率95.5%,纯度98.7%,具体反应如式ⅶ所示。
[0092]
制备得到的最终产物在氘代dmso中的核磁共振数据如下:1h[δ=2.80(2h,dd,j=13.9,5.8hz),3.71(2h,dd,j=13.9,1.5hz)4.93(2h,dd,j=5.8,1.5hz)]。
[0093]
在上述反应中,整体反应温度在0~5℃即可,温度过高会发生副反应。滴加的时间一般不少于1h,否则氯乙醛在水溶液中容易发生自聚合。由于硫氢化钠较为便宜,因此采用
少过量的硫氢化钠参加反应,有助于促进氯乙醛充分反应。
[0094]
实施例103,制备2,5-二羟基-1,4-二噻烷的制备方法,与实施例102的区别在于,硫氢化钠的水溶液中,硫氢化钠的质量分数为20%,产率为94.8%,纯度为98.5%。
[0095]
实施例104,制备2,5-二羟基-1,4-二噻烷的制备方法,与实施例102的区别在于,硫氢化钠的水溶液中,硫氢化钠的质量分数为30%,产率为93.4%,纯度为98.9%。
[0096]
实施例105,制备2,5-二羟基-1,4-二噻烷的制备方法,与实施例102的区别在于,硫氢化钠的水溶液中,硫氢化钠的质量分数为40%,产率为01.7%,纯度为98.7%。
[0097]
在实施例103~105中,硫氢化钠的物质的量均为2.1mol。
[0098]
由于水相氯乙醛制备完成后,体系中含有大量的盐酸,且会生成三氯乙烷的副产物,需要用大量小苏打中和盐酸,再进行上述反应。以山东某企业公开的环评资料显示制备1000公斤的2,5-二羟基-1,4-二噻烷,大约产生9800公斤的废水,三废处理压力较大,相较于本技术中的技术方案,成本和环境影响均有明显的不如。
[0099]
另外,设置对比例如下:对比例2,一种2,5-二羟基-1,4-二噻烷的制备方法,包括以下步骤:(1)调节氯乙醛ph称取1300kg氯乙醛水溶液a,缓慢添加小苏打调ph至3.5,溶液颜色为无色或浅黄色透明,得到氯乙醛水溶液b。
[0100]
氯乙醛水溶液a中,氯乙醛的质量含量为9-10%,游离酸的质量含量为9-10%,无色或浅黄色透明。
[0101]
(2)配制硫氢化钠溶液配制硫氢化钠溶液,质量浓度为32%,黄绿色透明,总量为360kg。
[0102]
(3)滴加部分硫氢化钠溶液称取200.0kg水加入反应釜中,滴加12kg硫氢化钠溶液,开启搅拌,冰水浴降温至8℃以下。
[0103]
(4)滴加反应等反应釜内温度降到5℃以下,开始滴加氯乙醛水溶液b和剩余硫氢化钠溶液,控制氯乙醛溶液b和硫氢化钠溶液的滴加速度比例为2:1;滴加过程中温度控制为≤25℃,每20分钟记录一次,反应2h。
[0104]
(5)熟化滴加结束后,熟化50分钟,温度控制≤25℃,出料。
[0105]
(6)抽滤、酸洗、水洗开启抽滤槽真空,尽量抽干母液,再用200kg工艺水和3.0kg盐酸的混合液洗涤、抽干;再用工艺水洗涤3次,每次工艺水用量为200kg,尽量抽干,洗至终点ph为6-7。
[0106]
(7)离心,烘干。
[0107]
在该方案中,2,5-二羟基-1,4-二噻烷的产率为90.0%,低于本技术中的技术方案,且反应过程中,每制备1000kg的2,5-二羟基-1,4-二噻烷会产生15600公斤的废水,废水量巨大,且调节好ph的氯乙醛b溶液的ph值依然达到3.5,在于硫氢化钠水溶液反应时不可避免的产生大量硫化氢,对于后续三废处理造成极大隐患。
[0108]
实施例106,制备2-氨基噻唑的制备方法,具体如下:配置含有质量分数为50%的硫脲水溶液167.5g(其中含硫脲1.1mol),向其中加入78.5(1mol)按照实施例97中的氯乙醛,并升温至70℃,保温并回流啊,4~6h后反应完毕,降至室温,加入氢氧化钠溶液调节至中性,进一步降温至0℃,过滤并用乙醇精制,得到2-氨基噻唑86.8g,产率86。7%,纯度98.8%,反应如式
ⅷ
所示。最终产物在氘代氯仿中的核磁共振
氢谱数据如下:1h nmrδ=7.05(1h,d,j=4.59,),6.49(1h,d,j=4.57),5.4(s,1h)。
[0109]
产品可以直接从水中析出,分离工艺较为简单。此处氢氧化钠换用氢氧化钾、碳酸氢钠均可,对反应产率无明显影响。
[0110]
本具体实施例仅仅是对本技术的解释,其并不是对本技术的限制,本领域技术人员在阅读完本说明书后可以根据需要对本实施例做出没有创造性贡献的修改,但只要在本技术的权利要求范围内都受到专利法的保护。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1