LCDR作为预防和/或治疗癌症的药物的治疗靶点中的应用的制作方法
lcdr作为预防和/或治疗癌症的药物的治疗靶点中的应用
技术领域
1.本发明涉及lcdr作为预防和/或治疗癌症的药物的治疗靶点中的应用,属于生物技术领域。
背景技术:
2.肿瘤是指生物体内由多种因素导致的其细胞异常生长而引发的疾病,按其对生物体的特性及危害又分为恶性肿瘤和良性肿瘤。恶性肿瘤,又称为癌症,有转移性和浸润性,良性肿瘤的特点是无浸润和转移的能力,常具有包膜,与正常组织分界清楚,生长缓慢,很少复发,对生物体影响小。
3.与正常细胞相比,肿瘤细胞通常具有以下特性:1、维持增殖信号;2、避免生长抑制;3、抵抗细胞凋亡;4、无限复制能力;5、诱导新生血管的生成;6、激活侵袭和转移;7、代谢重编程;8、逃逸免疫监控;9、基因组的不稳定性和易变性;10、引起炎症反应。目前,癌症已成为中国疾病死亡的主要因素之一,这主要是与肿瘤细胞区别于正常细胞的分子特性相关,使得对肿瘤的治疗成为世界性难题。
4.肺癌是是全世界范围内致死率及发病率最高的恶性肿瘤之一,具有高度的异质性,其在呼吸道各处形成,并可通过血管和淋巴管进行恶性扩张以及转移。根据目前最新的世界卫生组织(world healthorganization,who)病理学及组织学分类,将肺癌分为鳞状细胞癌,肺腺癌,小细胞癌以及肺神经内分泌肿瘤。
5.肺癌细胞获得的一系列的恶性特征往往是由细胞内基因突变所引起。通过全基因组测序揭示肺癌患者的癌细胞中存在着大量基因的突变以及染色体重排等。例如:膜表面的表皮生长因子受体(epidermalgrowth factor receptor,egfr)突变、alk(anaplastic lymphoma kinase,alk)重排、原癌基因1酪氨酸激酶(c-ros oncogene 1receptor kinase,c-ros,ros1)重排和braf v600e突变等,同时针对这些突变的靶向药在临床上已经取得了显著的疗效。
6.因此,找到更多与肺癌发生发展相关的分子机制对于肺癌治疗具有十分重要的临床应用价值。
技术实现要素:
7.为解决上述问题,本发明提供了一种分子标志物lcdr,所述分子标志物lcdr的核苷酸序列如seqid no:1所示。
8.本发明还提供了上述分子标志物lcdr,hnrnp k(核不均一核糖核蛋白k),上述分子标志物lcdr 和hnrnp k的复合物,和/或,laptm5(溶酶体相关跨膜蛋白5抗体)作为预防和/或治疗癌症的药物的治疗靶点中的应用。
9.在本发明的一种实施方式中,所述hnrnp k的氨基酸序列如seq id no:2所示。
10.在本发明的一种实施方式中,所述laptm5的氨基酸序列如seq id no:3所示。
11.在本发明的一种实施方式中,所述癌症为肺腺癌。
12.本发明还提供了抑制剂在制备预防和/或治疗癌症的药物中的应用,其特征在于,所述抑制剂具有如下至少一种用途:
13.(a)抑制细胞内上述分子标志物lcdr的表达;
14.(b)抑制细胞内hnrnp k的表达;
15.(c)抑制细胞内上述分子标志物lcdr和hnrnp k的复合;和/或,
16.(d)抑制细胞内laptm5的表达。
17.在本发明的一种实施方式中,所述hnrnp k的氨基酸序列如seq id no:2所示。
18.在本发明的一种实施方式中,所述laptm5的氨基酸序列如seq id no:3所示。
19.在本发明的一种实施方式中,所述癌症为肺腺癌;所述细胞为正常肺腺细胞和/或癌变的肺腺细胞。
20.本发明还提供了检测上述分子标志物lcdr,hnrnp k,上述分子标志物lcdr和hnrnp k的复合物,和/或,laptm5的检测试剂在制备用于诊断癌症的产品中的应用。
21.在本发明的一种实施方式中,所述hnrnp k的氨基酸序列如seq id no:2所示。
22.在本发明的一种实施方式中,所述laptm5的氨基酸序列如seq id no:3所示。
23.在本发明的一种实施方式中,所述检测权利要求1所述分子标志物lcdr的检测试剂包含用于扩增权利要求1所述分子标志物lcdr的特异性引物;
24.所述检测hnrnp k的检测试剂包含用于扩增编码所述hnrnp k的核苷酸序列的特异性引物;
25.所述检测laptm5的检测试剂包含用于扩增编码所述laptm5的核苷酸序列的特异性引物。
26.在本发明的一种实施方式中,所述用于扩增权利要求1所述分子标志物lcdr的特异性引物的核苷酸序列如seq id no:4以及seq id no:5所示。
27.在本发明的一种实施方式中,所述产品为检测试剂或检测试剂盒。
28.在本发明的一种实施方式中,所述癌症为肺腺癌。
29.本发明技术方案,具有如下优点:
30.本发明提供了分子标志物lcdr(核苷酸序列如seq id no:1所示),hnrnp k(核不均一核糖核蛋白k),分子标志物lcdr和hnrnp k的复合物,和/或,laptm5(溶酶体相关跨膜蛋白5抗体)作为预防和/或治疗癌症的药物的治疗靶点中的应用;研究表明:lcdr在肺腺癌组织中表达量相对较高;敲降lcdr发现肺腺癌细胞增殖能力显著下调且出现凋亡;敲降lcdr显著降低了肺腺癌细胞的肿瘤形成能力;lcdr通过形成二级结构,与hnrnp k相互结合,从而发挥其生物学功能,敲降hnrnp k出现了与lcdr一致的表型;lcdr与hnrnp k相互不影响表达量和亚细胞定位,同时在功能上也不能相互挽救;laptm5为lcdr/hnrnp k轴关键的下游基因,受到lcdr/hnrnp k的调控;laptm5具有与 lcdr/hnrnp k一致的作用,敲降laptm5出现了与lcdr/hnrnp k一致的表型;敲降lcdr或hnrnpk对肺腺癌细胞增殖以及克隆形成的抑制作用,均能通过过表达laptm5获得部分的挽救;laptm5的过表达可以使敲降lcdr或hnrnp k所导致的细胞凋亡得到部分的挽救;lcdr/hnrnp k/laptm5轴调控溶酶体介导的细胞死亡通路。
附图说明
31.图1:套式pcr的扩增程序。
32.图2:预跑胶的反应体系。
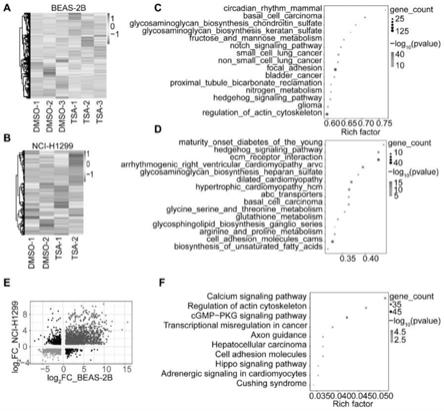
33.图3:乙酰化调控的基因谱以及通路富集。(a)tsa处理besa-2b差异表达基因的热图;(b) tsa处理nci-h1299差异表达基因的热图;(c)tsa处理besa-2b差异基因的通路富集;(d)tsa 处理nci-h1299差异基因的通路富集;(e)tsa处理besa-2b以及nci-h1299交集的差异基因的火山图;(f)tsa处理besa-2b以及nci-h1299交集的差异基因的通路富集。
34.图4:组蛋白乙酰化调控以及癌变过程差异变化的lncrnas的筛选。(a)tsa处理beas-2b、 nci-h1299以及7种癌种差异表达且同向变化lncrnas热图;(b)不同浓度tsa处理beas-2b, nci-h1299,hcc827细胞系h3k27ac以及h4k16ac蛋白水平的变化;(c)h3k27ac在lcdr、 slc12a5-as1、lucat1、nkila、mnx1-as1、mntn1-as1、snhk12的基因组上富集的峰图;(d) tsa处理beas-2b、nci-h1299以及7种癌种差异表达且同向变化并受到h3k27ac修饰的lncrnas 热图;(e-g)qrt-pcr检测不同浓度tsa处理beas-2b(e),nci-h1299(f),hcc827(g)细胞系,lcdr转录本水平的变化;(h-j)qrt-pcr检测不同浓度tsa处理beas-2b(h),nci-h1299 (i),hcc827(j)细胞系h3k27ac对lcdr启动子的富集效率。
35.图5:lcdr全长克隆以及相对表达量检测。(a)lcdr 3
′
race和5
′
race分析,pcr产物测序图(左)与5
′
和3
′
race pcr产物的凝胶电泳图(右);(b)lcdr的northern blot分析;(c) lcdr在人类基因组grch38上的定位;(d)qrt-pcr检测lcdr在肺癌细胞系里的转录本水平;(e) qrt-pcr检测lcdr在肺癌细胞系里的分子数。双尾t检验分析组间的差异,数据表示为mean
±
sem。
36.图6:(a)lcdr编码能力的预测;(b)lcdr启动子转录活性的分析;(c)lcdr保守性分析。
37.图7:lcdr分子的亚细胞定位。(a)lcdr分子fish图红色表示lcdr的杂交信号,蓝色表示dapi染料信号;(b)。qrt-pcr检测lcdr在细胞质与核的定位比例。使用gapdh蛋白与h3 组蛋白分别作为细胞质与核蛋白质的阳性对照,使用gapdh与u1基因分别作为细胞质与核基因的阳性对照,wcl、cyto与nuc分别表示全细胞,细胞质与细胞核裂解物。数据表示为mean
±
sem。
38.图8:c-jun调控lcdr的转录。(a)qrt-pcr检测敲降c-jun lcdr转录水平变化情况;(b) qrt-pcr检测过表c-jun lcdr转录水平变化情况;(c)gse92783数据集的chip测序数据,c-jun 在a549细胞系中lcdr启动子区域上的富集水平;(d)生物信息预测c-jun在lcdr启动子区域的结合位点;(e)chip验证c-jun结合在lcdr的启动子区域;(f)c-jun与lcdr结合位点的插画突变示意图;(g)双酶报试验验证c-jun与lcdr启动子的相互结合;(h)chip实验验证tsa处理对于c-jun富集lcdr启动子的影响。双尾t检验分析组间的差异,数据表示为mean
±
sem。
39.图9:lcdr表达量的改变。(a)qrt-pcr检测nci-h1299细胞系中sh-lcdr的敲降效率;(b) qrt-pcr检测nci-h1299细胞系中si-lcdr的敲降效率;(c)qrt-pcr检测nci-h1299细胞系中lcdr 的过表倍数。双尾t检验分析组间的差异,数据表示为mean
±
sem。
40.图10:lcdr表达量的改变对nci-h1299增殖活力的影响。(a)shrna系统敲降lcdr的表达,对nci-h1299增殖活力的影响;(b)sirna系统敲降lcdr的表达,对nci-h1299增殖活力
的影响; (c)过表lcdr对nci-h1299增殖活力的影响。双尾t检验分析组间的差异,数据表示为mean
±
sem。
41.图11:lcdr表达量的改变对nci-h1299克隆形成的影响。(a)shrna系统敲降lcdr的表达,对nci-h1299克隆形成的影响;(b)sirna系统敲降lcdr的表达,对nci-h1299克隆形成的影响; (c)过表lcdr对nci-h1299克隆形成的影响。双尾t检验分析组间的差异,数据表示为mean
±
sem。
42.图12:敲降lcdr诱导细胞的凋亡。(a)shrna系统敲降lcdr表达nci-h1299的光镜代表图; (b)sirna系统敲降lcdr表达nci-h1299的光镜代表图;(c)sh-lcdr流式检测nci-h1299细胞凋亡代表图;(d)sh-lcdr流式检测nci-h1299细胞凋亡比例统计图;(e)si-lcdr流式检测 nci-h1299细胞凋亡代表图;(f)si-lcdr流式检测nci-h1299细胞凋亡比例统计图。双尾t检验分析组间的差异,数据表示为mean
±
sem。
43.图13:敲降lcdr诱导细胞的凋亡。(a)shrna系统敲降lcdr表达cytochrome c定位代表图; (b)sirna系统敲降lcdr表达cytochrome c定位代表图;(c)shrna系统敲降lcdr表达caspase, parp蛋白质表达情况;(d)sirna系统敲降lcdr表达caspase,parp蛋白质表达情况。
44.图14:敲降lcdr对nci-h1299肿瘤形成能力的影响。(a)敲降lcdr抑制nci-h1299的肿瘤的增殖能力;(b)敲降lcdr抑制nci-h1299的成瘤能力;(c)敲降lcdr的nci-h1299细胞抑制肿瘤生长;(d)敲降lcdr免疫组化染色cleaved caspase3代表图;(e)免疫组化cleaved caspase3 免疫组化得分统计。双尾t检验分析组间的差异,数据表示为mean
±
sem。
45.图15:lcdr与hnrnp k特异性结合。(a)lcdr rna pμll down银染胶图;(b)western blot 验证lcdr与hnrnp k特异性结合;(c)rip实验验证hnrnp k与lcdr的相互结合,igg为阴性对照;(d)lcdr原位杂交和hnrnp k免疫荧光验证lcdr和hnrnp k在细胞核内共存。双尾t检验分析组间差异,数据表示为mean
±
sem。
46.图16:hnrnp k结合lcdr位点的鉴定。(a)western blot检测全长以及截短型的lcdr与hnrnpk的相互作用;(b)rnafold在线软件对lcdr二级结构的预测;(c)lcdr与hnrnp k潜在结合位点突变示意图;(d)western blot检测lcdr,lcdr(1-500)以及mut-lcdr与hnrnp k的相互作用;(e)emsa鉴定lcdr与hnrnp k的相互结合;(f)rip-qpcr检测hnrnp k与lcdr以及 mut-lcdr的相互结合。双尾t检验分析组间差异,数据表示为mean
±
sem。
47.图17:lcdr结合hnrnp k结构域的鉴定。(a)hnrnp k截短体示意图;(b)western blot检测hnrnp k截短体的蛋白表达情况;(c)rip-qpcr检测全长以及截短体的hnrnp k对lcdr的富集水平。双尾t检验分析组间差异,数据表示为mean
±
sem。
48.图18:hnrnp k表达量的改变。(a)qrt-pcr检测nci-h1299细胞系中hnrnp k的敲降效率; (b)qrt-pcr检测nci-h1299细胞系中hnrnp k的过表倍数。双尾t检验分析组间差异,数据表示为mean
±
sem。
49.图19:hnrnp k对细胞增殖的影响。(a)敲降hnrnp k对nci-h1299增殖活力的影响;(b) 过表hnrnp k对nci-h1299增殖活力的影响。双尾t检验分析组间的差异,数据表示为mean
±
sem。
50.图20:hnrnp k对细胞克隆形成能力的影响。(a)敲降hnrnp k对nci-h1299克隆形成能力的影响;(b)过表hnrnp k对nci-h1299克隆形成能力的影响。双尾t检验分析组间的
差异,数据表示为mean
±
sem。
51.图21:hnrnp k对细胞死亡的影响。(a)敲降hnrnp k细胞光镜代表图;(b)敲降hnrnp k 细胞凋亡流式代表图;(c)敲降hnrnp k细胞凋亡流式统计图;(d)敲降hnrnp k免疫荧光检 cytochrome c的定位;(e)敲降hnrnp k western blot检测caspase,parp蛋白质表达情况。双尾t 检验分析组间的差异,数据表示为mean
±
sem。
52.图22:lcdr与hnrnp k相互关系。(a)lcdr和hnrnp k相互结合的免疫荧光图,蓝色表示的是dapi的信号,绿色表示的是hnrnp k蛋白的信号,红色表示的是lcdr的信号;(b)qpcr检测敲降lcdr,hnrnp k mrna水平变化情况;(c)western blot检测敲降lcdr,hnrnp k蛋白水平变化情况;(d)qpcr检测敲降hnrnp k,lcdr转录本变化情况;(e)western blot检测敲降hnrnpk,hnrnp k蛋白水平变化情况。双尾t检验分析组间差异,数据表示为mean
±
sem。
53.图23:lcdr与hnrnp k功能上的挽救。(a)过表hnrnp k细胞株敲降lcdr的细胞增殖曲线;(b)过表lcdr细胞株敲降hnrnp k的细胞增殖曲线。双尾t检验分析组间差异,数据表示为 mean
±
sem。
54.图24:lcdr与hnrnp k对细胞凋亡相关基因的表达的影响。(a-b)qrt-pcr检测敲降lcdr (a)以及hnrnp k(b)凋亡相关基因mrna变化水平;(c-d)western blot检测敲降lcdr(c) 以及hnrnp k(d)凋亡相关基因蛋白水平的变化。双尾t检验分析组间差异,数据表示为mean
±
sem。
55.图25:lcdr与hnrnp k共同调控溶酶体通路。(a-b)火山图表示敲降lcdr(a)以及hnrnpk(b)差异表达的基因,红色代表上调的基因,绿色代表下调的基因;(c-d)敲降lcdr(c)以及 hnrnp k(d)差异表达的基因的通路富集;(e)敲降lcdr,hnrnp k交集差异基因的韦恩图;(f) 敲降lcdr,hnrnp k交集差异基因的通路富集;(g)敲降lcdr,hnrnp k交集差异基因的的kegg 通路富集;(h-i)敲降lcdr,hnrnp k差异基因溶酶体通路热图;(j-k)qrt-pcr检测敲降lcdr (j)以及hnrnp k(k)溶酶体通路基因的mrna水平的变化。双尾t检验分析组间差异,数据表示为mean
±
sem。
56.图26:lcdr和hnrnp k调控laptm5的表达。(a)western blot检测敲降lcdr,laptm5蛋白水平的变化;(b)western blot检测敲降hnrnp k,laptm5蛋白水平的变化;过表lcdr,laptm5mrna以及蛋白水平的变化(c-d);过表hnrnp k,laptm5 mrna以及蛋白水平的变化(e-f); (g)laptm5与lamp1免疫荧光共定位代表图;(h)过表达laptm5与lamp1免疫荧光共定位代表图。
57.图27:laptm5表达量的改变。(a)qrt-pcr检测nci-h1299细胞系中laptm5的敲降效率; (b)qrt-pcr检测nci-h1299细胞系中laptm5的过表倍数。双尾t检验分析组间差异,数据表示为mean
±
sem。
58.图28:laptm5对细胞增殖的影响。(a)敲降laptm5对nci-h1299增殖活力的影响;(b) 过表laptm5对nci-h1299增殖活力的影响。双尾t检验分析组间的差异,数据表示为mean
±
sem。
59.图29:laptm5对细胞克隆形成能力的影响。(a)敲降laptm5对nci-h1299克隆形成能力的影响;(b)过表laptm5对nci-h1299克隆形成能力的影响。双尾t检验分析组间的差异,数据表示为mean
±
sem。
60.图30:laptm5对细胞死亡的影响。(a)敲降laptm5细胞光镜代表图;(b)敲降laptm5 细胞凋亡流式代表图;(c)敲降laptm5细胞凋亡流式统计图;(d)敲降laptm5 cytochrome c 的定位;(e)敲降laptm5 western blot检测caspase,parp蛋白质表达情况。双尾t检验分析组间的差异,数据表示为mean
±
sem。
61.图31:lcdr、hnrnp k和laptm5调控溶酶体膜通透性。(a-b)lysotracker、lamp1、ctsb 的免疫荧光染色代表图;(c)扫描电镜观察溶酶体膜的完整性,红色箭头代表膜破裂的位点。
62.图32:lcdr与hnrnp k通过laptm5调控细胞死亡。稳定过表laptm5的细胞系敲降lcdr (a)和hnrnp k(c)细胞增殖能力的检测;稳定过表laptm5的细胞系敲降lcdr(b)和hnrnpk(d)细胞克隆形成能力的评估;稳定过表laptm5的细胞系敲降lcdr(e-f)和hnrnp k(g-h) 细胞凋亡的评估;ca-074me处理细胞敲降lcdr(i-j)、hnrnp k(k-l)和laptm5(m-n)细胞凋亡的评估。双尾t检验分析组间差异,数据表示为mean
±
sem。
63.图33:lcdr与hnrnp k通过laptm5调控细胞死亡。(a)稳定过表laptm5的细胞株再敲减 lcdr以及hnrnp k肿瘤的生长曲线;(b)稳定过表laptm5的细胞株再敲减lcdr以及hnrnp k 小鼠皮下成瘤能力可得到部分挽救;(c)稳定过表laptm5的细胞株再敲减lcdr以及hnrnp k肿瘤重量的统计图。双尾t检验分析组间差异,数据表示为mean
±
sem。
64.图34:稳定过表laptm5的细胞株再敲减lcdr以及hnrnp k免疫组化(a)代表图以及cleavedcaspase 3得分(b)统计图。双尾t检验分析组间差异,数据表示为mean
±
sem。
65.图35:lcdr和hnrnp k结合在laptm5-3
′
utr。(a)hnrnp k rip seq数据分析hnrnp k peak 在laptm5转录本上的分布;(b)lcdr rna-seq、hnrnp k rna-seq、hnrnp k rna-seq以及hnrnpk-eclip交集基因韦恩图;(c)lcdr rna-seq、hnrnp k rna-seq、hnrnp k rip-seq以及hnrnpk-eclip交集基因kegg通路富集;(d)双酶报实验检测hnrnp k与laptm5编码区以及3
′
utr 的
·
相互结合;(e-f)laptm5-3
′
utr与hnrnp k潜在结合位点(e)以及突变(f)示意图;(g) rna pμll down实验验证hnrnp k与laptm5-3
′
utr的相互结合;(h)rip-qpcr检测hnrnp k与 laptm5-3
′
utr以及laptm5-3
′
utr的相互结合;(i)emsa鉴定hnrnp k与laptm5-3
′
utr的相互结合;(j)rip-qpcr检测全长以及截短体的hnrnp k对laptm5的富集水平。双尾t检验分析组间的差异,数据表示为mean
±
sem。
66.图36:lcdr和hnrnp k调控laptm5 mrna的稳定性。(a)敲降lcdr过表达hnrnp k,laptm5转录本稳定性的检测;(b)敲降hnrnp k的过表达lcdr,laptm5转录本稳定性的检测。
67.(c)qrt-pcr检测敲降hnrnp k过表lcdr,laptm5转录本水平的变化情况;(d)qrt-pcr检测敲降lcdr过表hnrnp k,laptm5转录本水平的变化情况(e-f)双酶报实验鉴定lcdr(e)以及 hnrnp k(f)与laptm5-3
′
utr的相互结合。双尾t检验分析组间的差异,数据表示为mean
±
sem。双尾t检验分析组间的差异,数据表示为mean
±
sem。
68.图37:lcdr和hnrnp k协同促进阅读laptm5。(a)rip-qpcr检测敲降lcdr的细胞系中 hnrnp k富集laptm5的水平(b)rip-qpcr检测过表lcdr的细胞系中hnrnp k富集laptm5的水平;(c)pμll down检测敲降hnrnp k细胞系中,lcdr富集laptm5的水平;(d)pμll down 检测过表hnrnp k细胞系中,lcdr富集laptm5的水平;(e)qpcr检测使用蛋白酶k处理rna pμlldown样品前后,生物素标记lcdr对laptm5的结合水平;(f)hnrnp k与lcdr以及laptm5 结合位点突变示意图;(g)rip-qpcr检测双过表lcdr和hnrnp k的细胞系中hnrnp k富集
laptm5 的水平;(h)双酶报实验检测双过表lcdr和hnrnp k与laptm5-3
′
utr共转,对酶报活性的影响。双尾t检验分析组间差异,数据表示为mean
±
sem。
69.图38:lcdr促进hnrnp k结合laptm5。(a)emsa实验检测lcdr和laptm5-3
′
utr与 hnrnp k蛋白孵育,对hnrnp k蛋白迁移速率的影响;(b-c)emsa实验检测laptm5-3
′
utr对hnrnpk蛋白和lcdr结合的影响;(d-e)emsa实验检测lcdr对hnrnp k蛋白和laptm5-3
′
utr结合的影响。双尾t检验分析组间差异,数据表示为mean
±
sem。
70.图39:lcdr,hnrnp k以及laptm5在肺腺癌组织中表达情况。(a)lcdr原位杂交癌与癌旁表达情况代表图;(b)lcdr原位杂交表达量统计图;(c)hnrnp k免疫组化癌与癌旁表达情况代表图;(d)hnrnp k免疫组化表达量统计图;(e)laptm5免疫组化癌与癌旁表达情况代表图;(f) laptm5免疫组化表达量统计图。
71.图40:lcdr,hnrnp k以及laptm5相关相以及roc分析。(a)lcdr与hnrnp k相关性分析;(b)lcdr与laptm5相关性分析;(c)hnrnp k与laptm5相关性分析;(d)lcdr,hnrnpk以及laptm5 roc联合分析。
具体实施方式
72.提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
73.下述实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
74.实施例1:一种分子标志物
75.本实施例提供了一种分子标志物lcdr,所述分子标志物lcdr的核苷酸序列如seq id no:1所示。
76.实验例1:分子标志物lcdr准确转录本的获取及分子标志物lcdr的功能验证
77.(一)实验材料与方法
78.1基因克隆
79.1.1pcr
80.从cdna或者质粒中克隆基因时,为了确保克隆基因的高保真性,采用fast pfu dna polymerase (transgen)。反应体系见表1。
81.表1质粒构建的pcr反应体系
82.83.注意事项:1)反应体系的模板不要过量。以cdna为模板配置50μl反应体系为例,其模板量一般在50~100ng为宜;2)根据引物tm值设定退火温度反应时,若出现杂带,可以提高退火温度以消除,最高可至72℃;若无条带,则可以降低退火温度,直至出现条带;3)注意根据产物大小和酶的扩增速度调整合适的延伸反应时间(一般1分钟能延伸1kb)。
84.1.2dna凝胶电泳
85.采用琼脂糖凝胶电泳。加入gelred染料(gelred核酸染料biotium41003,用水溶解,每50ml琼脂糖溶液中加入5μl gelred 10,000
×
储液)的2%(g/100ml)的琼脂糖凝胶,在120v的电压下进行电泳,电泳结果可直接在紫外下观察。
86.常用的电泳缓冲液为tae,提前配置50x的储存液,使用时用去离子水稀释至1x。配置体积为1l 的50x tae需要tris 242g,0.5m的edta(ph8.0)100ml和冰醋酸57.1ml。
87.注意事项:1)根据所需样品量以及用途,选择配置溶液的体积,以及合适的梳子和溶液浓度;2) 注意根据目的条带大小选择合适的marker;3)注意dna的移动方向是由负极到正极。
88.电泳步骤:
①
用1
×
tae-buffer按照被分离dna分子的大小配制1%的琼脂糖凝胶50ml(称量0.5g 琼脂,加入50ml 1
×
tae),在微波炉中加热至溶解;
②
待溶液冷却至50℃,加入gelred核酸染料,充分混匀,倒入适宜的已插好梳子的胶模中;
③
在凝胶完全凝固后,取走梳子,将凝胶转移至装有1
×
tae 缓冲液的电泳槽中,使缓冲液没过胶面;
④
dna样品与10
×
loading-buffe混合后,取混合物加至样品孔中,并加入5μl dna ladder;
⑤
通电,使dna向阳极(红线)移动;
⑥
在指示条带移动适当距离后切断电流,取出凝胶,在紫外灯下观察凝胶。
89.1.3(切胶)回收
90.对于只有特异条带的反应(pcr或酶切等)产物,可以直接回收;而对于存在非特异条带的产物,则需要切下特异条带位置的凝胶后进行回收。
91.注意:1)切胶需要在紫外灯下进行,为了防护安全,注意使用遮光板;切下的条带要尽可能剔除无用区域,以提高回收效率;2)回收时,为了加速胶块溶解可以将其切碎;吸附柱容积为800μl,若样品体积大于800μl可分批加入;为了增加洗脱效率,可以将洗脱液加热至65~70℃。
92.凝胶回收简要步骤(天根琼脂糖凝胶回收试剂盒):
①
向吸附柱cb2中加入500μl的平衡液bl, 12000rpm离心1min,倒掉收集管中的废液;
②
向胶块中加入等体积溶液pc(如果凝胶中0.1g,其体积可视为100μl,则加入100μl pc溶液),50℃水浴放置10min左右,待胶全部溶解;
③
将所得溶液加入一个吸附柱cb2中,12000rpm离心1min,倒掉收集管中的废液;
④
向吸附柱cb2中加入600μl漂洗液pw(检查是否已加入无水乙醇),12000rpm离心1min,倒掉收集管中的废液;
⑤
重复操作步骤
④
;
⑥
将吸附柱cb2放回收集管中,12000rpm离心2min,将吸附柱cb2在超净台中风干放置10min;
⑦
将吸附柱cb2放入一个干净的离心管中,加入50μl ddh2o,在室温中放置2min后,12000rpm离心2min,收集dna溶液。
93.注意事项:对于pcr反应产物回收,只有步骤2和3有差异。即可以直接加入等体积的pc溶液,混匀后加入到吸附柱cb2中进行回收。
94.1.4酶切
95.以psih-h1-puro质粒为例,假设其浓度为1000ng/μl,酶切1μg,体系20μl,即:
[0096][0097]
注意事项:1)酶的总体积最大不能超过总体系的10%;2)对于切下片段较大而导致直接回收时不仅会收集到目的片段的情况,建议反应时使用fastdigestgreenbuffer;3)鉴于一些酶可能放置时间过久,效率降低,因此建议酶切时间适当延长至1.5~2h。
[0098]
1.5与载体连接
[0099]
反应体系见表2。
[0100]
表2反应体系
[0101]
试剂用量2x t4 dna ligase buffer5μlvector dna(已酶切)50nginsert dna(已酶切)xt4 dna ligase1μlh2o补足到10μl
[0102]
注意事项:1)insertdna的量根据它和vectordna的长度和浓度确定,通常摩尔比insertdna/vectordna为5-10;其摩尔数简单概括为质量/bp数。即以一个10μl的反应体系为例,假设vectordna浓度为50ng/μl,长度为8000bp;insertdna浓度为100ng/μl,长度为1000;摩尔比insertdna/vectordna设定为10,则所需的insertdna的体积为x,其计算公式为:(100x/1000)/(50/8000)=10,则x=5/8μl;
[0103]
2)连接反应一般室温2h左右即可完成,为了增加连接效率,可以四度过夜反应,效果更佳。
[0104]
1.6转化、涂板
[0105]
转化需要用到的液态lb培养基与涂板用到的固态lb培养基都需要提前配置好。1l液态lb培养基配置,需要胰化蛋白胨10g、酵母提取物5g、nacl10g、加入950ml去离子水溶解,用5m的naoh调节ph至7.0,后,定容至1l。之后121℃20min高压灭菌。
[0106]
固态培养基则需另外加入15~20g琼脂粉。另外灭菌时需要设定60℃保温,以免其凝固。取出后,用流水降温至手背触摸底部不烫时,于超净台中1:1000加入相应抗生素,摇匀后倒平板。10ml倒一个平板。平板倒入培养基后,摇匀,半开盖子于超净台中开风降至室温后,用封口膜封口后,装袋保存于4℃备用。
[0107]
转化步骤:
①
从-80℃冰箱取出感受态细胞,放在冰上溶解;
②
每管感受态包含100μl,一次反应50μl足以。将连接产物加入50μl感受态中,冰上放置15min。然后42℃热激90s,置于冰上3min;
③
每管中加入200μl无抗的lb,37℃200rpm摇菌30~60min。
[0108]
注意事项:避免手碰到感受态的管底而影响其状态。
[0109]
涂平板步骤:
①
玻璃涂抹器头部在95%乙醇中浸泡后,于酒精灯火焰灭菌后,置于支架上降温。每涂抹完一次需重复该步骤;
②
从4℃取出所需平板,加入适量的转化菌液,用涂抹器涂抹均匀。倒置于37℃细菌培养箱过夜。
[0110]
注意事项:1)上述操作需要在超净工作台中进行;2)为了避免平板中长出太多克隆或者没长克隆,每个转化菌株可以分别涂两块平板:1/10菌液涂布一个平板,剩余的涂布在另一平板上;3)最好在连接时做一个只包含vector dna(已酶切)的阴性组,以确认酶切是否干净。
[0111]
1.7挑克隆和菌液pcr:
[0112]
平板培养12小时后,从细菌培养箱中取出,确认有无克隆。
[0113]
挑克隆和菌液pcr步骤:
①
按照准备挑的克隆数,配置pcr反应体系,不用预留模板的体积。
②
取出灭菌的ep管,加入500μl已按照1:1000加有相应抗生素的lb培养基;镊子在酒精灯火焰上灼烧灭菌后,稍微晾凉后,夹取20μl枪头,挑取平板上的单克隆,在相应的加好pcr反应体系的pcr管中蘸几次后,放入相应的ep管中。
③
重复操作
②
直至挑完克隆。pcr反应混匀、离心后,于pcr仪反应,通过电泳拍照确认克隆是否为阳性;ep管则与细菌摇床37℃200rpm摇菌。
④
确认阳性克隆后,从相应 ep管中吸取100μl至新的ep管中送测序。
[0114]
注意事项:1)上述操作需要在超净工作台中进行;2)一个平板挑取4~6个克隆进行测序。
[0115]
2细胞培养
[0116]
2.1实验材料
[0117]
本研究所使用细胞系及其所用培养基名称见表3。所有细胞均购买自中国科学院上海细胞库。所有培养基及胎牛血清(fetal bovine serum,fbs)、青链霉素混合液(penicillin-streptomycin solution,ps)和 0.25%胰蛋白酶溶液购买自gibco公司。
[0118]
表3细胞系及培养基
[0119]
细胞系及培养基来源beas-2b上海细胞库nci-h1299上海细胞库lenti-x
tm
293t上海细胞库dmemgibcorpmi-1640gibco
[0120]
2.2实验方法
[0121]
(1)细胞复苏
[0122]
准备复苏细胞之前先确认恒温水浴锅的温度是37℃,打开液氮罐,取出预先冻存的细胞在水浴锅里摇晃,使细胞快速融化,然后1000rpm、5min离心,离心期间取出10cm皿,加培养基,离心结束后,取出细胞取出上清,加入1ml由10vt%fbs、1vt%ps和培养基组成的细胞培养基(完全培养基)重悬细胞,加入细胞培养皿,晃动培养皿,使细胞均匀分布,放置于细胞培养箱中培养。
[0123]
(2)细胞传代
[0124]
待细胞长到90%密度时,从培养箱里移出细胞到超净台(预先紫外照射30min),弃掉上清,取2ml1
×
pbs对细胞进行洗涤两遍,1ml 0.25%的胰酶消化2min后,用2ml完全培养终消化,用移液器慢慢吹打细胞,使细胞从培养皿上脱落,再移至15ml离心管,1000rpm、5min离心,离心期间取出所需10cm 皿个数,加培养基,离心结束后,取出细胞取出上清,加入1ml由10vt%fbs、1vt%ps和培养基组成的细胞培养基(完全培养基)重悬细胞,加入细胞
培养皿,慢慢晃动培养皿,使细胞均匀分布,放置于细胞培养箱中培养。
[0125]
(3)细胞冻存
[0126]
在细胞传代离心过程中,准备细胞冻存液,按照细胞培养基:fbs:dmso=6:3:1比例配制,待离心结束后,去除上清,加入1ml细胞冻存液吹打混匀,再转移到细胞冻存管中,标好细胞名称,冻存时间,传代次数,用棉花包裹放入-80℃冻存,一个星期后移到液氮罐中。
[0127]
3质粒提取
[0128]
质粒提取步骤:
①
取15ml培养过夜(16小时)的菌液至15ml的离心管中,12000rpm离心1分钟收集细菌团块;
②
向留有菌体沉淀的离心管中加入500μl buffer p1,使用涡旋振荡器或者移液枪充分混匀;
③
向离心管中加入500μl buffer p2,轻柔地上下颠倒混匀11次,使菌体得到充分的裂解,放置于室温6分钟,此时离心管中的融汇会逐渐变得清澈透明;
④
向离心管中缓慢加入500μl buffer e3,随即上下颠倒混匀11次,此时离心管中会逐渐出现白色絮状的沉淀,放置于室温(25℃)5分钟。12000rpm 离心5分钟,收集上清,将得到的上清加入过滤柱中,放置1分钟,12000rpm离心1分钟;
⑤
根据得到的滤液的体积加入相应的异丙醇,然后上下颠倒混匀,室温放置10分钟;
⑥
向吸附柱中加入200μl bufferps,12000rpm离心2分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中;
⑦
将上述滤液与异丙醇的混合溶液加入到平衡好的吸附柱中;
⑧
室温放置1分钟,12000rpm离心1分钟,倒掉收集管中的滤液,直至所有的液体过柱完成;
⑨
向吸附柱中加入750μl buffer pw,室温(25℃)放置2分钟,12000rpm 离心1分钟,倒掉收集管中的废液,重复漂洗2次;
⑩
将吸附柱换一个新的1.5ml的离心管,并在超净台中吹风放置10min;向吸附膜的中间部位加入100μl无菌水,室温放置5分钟,12000rpm离心2分钟。将得到的溶液重新加入到吸附柱中,室温放置5分钟,12000rpm离心2分钟;使用nanodrop测定质粒浓度,于-20℃冰箱保存质粒。
[0129]
4慢病毒包装与感染
[0130]
准备10cm皿的293t细胞,待细胞贴壁密度为60%。质粒转染体系见表4~5。
[0131]
表4 a管转染体系
[0132]
试剂用量pei reagent25.5μl(总质粒3倍)opti-memup to 100μl
[0133]
表5 b管转染体系
[0134][0135]
慢病毒包装与感染步骤:
①
将配置好的a、b两个ep管混匀,静置15分钟;
②
取出293t,吸去上清,加入4ml无血清dmem/10cm皿,将
①
的混合液加入293t,放置6h后更换完全培养基;
③
收取不同时间点上清(24h,48h),4500rpm离心15min,取离心后上清加入到目的细胞中;
④
反复感染两次即可,为了获得稳定细胞株,可以进行相应的筛选。
inhibitorcocktai)重悬细胞核沉淀,沿着管壁缓慢的加入500μl的buffers2(0.35mm蔗糖、0.5mmmgcl2、1%proteaseinhibitorcocktai)。使用4℃离心机3500rpm离心10min,弃上清加入200μlripa强裂解液充分混匀,冰上静置30min,使用4℃离心机12000rpm离心15min,吸取上清用于细胞核蛋白以及rna的提取。
[0149]
7细胞增殖与克隆形成
[0150]
7.1细胞增殖
[0151]
细胞基因敲除或者过表达之后,为了进一步研究基因的表达量对细胞功能的影响,使用celltiter-gloluminescent
tm
cellviabilityassayassays(ctg)进行检测96板中细胞的相对个数,具体步骤如下:
[0152]
(1)收集细胞:将胰酶覆盖细胞培养皿的表面,待细胞消化完成加入完全培养基终止消化,使用移液枪缓慢的吹打细胞并将细胞转移至15ml的离心管中,1000rpm离心5min,使用移液泵吸弃上清,加入1ml的完全培养基悬浮细胞沉淀,并吹打混匀。
[0153]
(2)细胞计数:根据细胞沉淀,进行相应的稀释。吸取上述步骤的细胞悬浮液20μl于干净无菌的ep管中,加入180μlpbs缓慢上下颠倒混匀,此时细胞浓度稀释至至原来的1/10。吸取10μl稀释过后的细胞悬浮液沿着技术板的加入孔缓慢加入细胞悬液,直至全部覆盖,同时尽量不要产生气泡。
[0154]
(3)细胞铺板:根据计数的结果,加入完全培养基或者细胞悬液,最终100μl的细胞悬浮液包含1000~2000个细胞即可,使用移液枪准确将细胞悬液加入到96孔板中,使用ctg进行读数,即为第一天的细胞数。
[0155]
(4)细胞读数:读取3组数据,每间隔一天读取一次。
[0156]
7.2克隆形成
[0157]
克隆形成步骤:
①
吸取细胞增殖的悬浮液,加入到6孔板中,细胞数为500~1500个,加入完全培养基2ml,放置于37℃、5%(v/v)co2细胞培养箱中进行培养,经常隔天观察,培养周期为2周;
②
当观察到6孔板中出现大小以及数目适当的细胞克隆时,终止培养,取出6孔板,pbs清洗3次,加入4%(g/100ml)的多聚甲醛室温固定10min;
③
吸弃4%的多聚甲醛,pbs再次洗涤3次,加入结晶紫室温放置1h进行染色;
④
待染色完成,回收结晶紫,染着6孔板的沿壁缓慢的清洗,直至清洗的水流变得清亮。
[0158]
8rna提取与逆转录
[0159]
8.1rna提取
[0160]
rna提取步骤:
[0161]
①
细胞或组织加入trizol后,室温(25℃)放置5min使其充分裂解,这会让核酸蛋白复合物完全分离(可轻轻翻转离心管混匀);
[0162]
②
4℃离心机12000rpm离心10min,后吸取上清转入新的rnasefree的ep管中。ep管需作好标识,同时注意防止标记被有机溶剂侵蚀;
[0163]
③
加入氯仿进行抽提,以去除蔗糖、蛋白等杂质,促进水相与有机相的分离。每1mltrizol至少加入0.2ml氯仿(1/5体积)。盖紧样品瓶盖,用手用力摇晃试管15s使其充分混匀,室温静置5min;
[0164]
④
加入异丙醇沉淀水相rna。加入氯仿静置完成后,4℃离心机12000rpm离心20min,此时分相为三层。小心地将上层水相转入新的1.5ml无rna酶离心管中(约400-500μ
l),再加入等体积的异丙醇(按照0.5ml异丙醇/mltrizol加入),混匀后,室温放置10min或者-20℃中1小时;
[0165]
⑤
4℃离心机12000rpm离心10min,小心弃上清。(离心前rna沉淀经常看不见,离心后在管侧或管壁形成胶状沉淀);
[0166]
⑥
加入75vt%乙醇洗涤rna沉淀,以使rna沉淀中的盐离子被充分溶解。加入1ml75vt%乙醇,将rna沉淀弹起漂洗。每使用1mltrizol则至少使用1ml75vt%乙醇对rna沉淀进行洗涤;
[0167]
⑦
rna沉淀洗涤后,4℃离心机12000rpm离心10min。吸弃上清,注意不要倒出沉淀,剩余少量液体短暂离心后用枪头吸出,注意不要吸弃沉淀;
[0168]
⑧
将ep管打开置于超净台吹干(15min)。rna沉淀不能吹太干燥,因其完全干燥后很难溶解,但又要充分避免乙醇影响后续试验;
[0169]
⑨
根据试验需要以及rna沉淀多少,加入depc水(100μl),反复吹打混匀,充分溶解rna;
[0170]
⑩
定量(nanodrop):用紫外分光光度仪检测所提rna,通过od
260/280
来检测rna纯度及浓度;
[0171]
其中,od
260/280
作为参考值:
[0172]
od
260/280
在1.9~2.1之间,可以认为rna的纯度较好,浓度:μg/μl=od
260
*40*稀释倍数/1000;
[0173]
od
260/280
值小于1.8,则表明蛋白杂质较多;
[0174]
od
260/280
值大于2.2,则表明rna部分降解;
[0175]
od
260/280
值小于2.0,则表明裂解液中有异硫氰酸胍和β-巯基乙醇法残留。
[0176]
8.2逆转录
[0177]
逆转录步骤:
[0178]
(1)计算所需rna量:由于去除基因组dna(gdna)反应中对rna所需量只需1μg,故需对rna进行定量计算。即rna所需量=1000/所提rna浓度。
[0179]
(2)去除基因组dna(体系:10μl):将试剂放置于冰上或冰盒中,按表6体系(该体系仅为一份样品体系)配置反应液总管,最后分装于pcr管中,根据计算结果加入rna样品并作好标记。配置完成后,混匀,将pcr管放于pcr仪,按表7程序进行反应。
[0180]
表6pcr体系
[0181]
2.5
×
gdna eraser buffer2μl1.gdna eraser1μlrna1μg(rna所需量=1000/所提rna浓度)6.rnase free dh2o(h2o=7-1000/所提rna浓度)μl
[0182]
表7pcr程序
[0183]
42℃2min4℃保存
[0184]
(3)反转录(体系:10μl):在样品进行去除gdna反应的同时,按表8体系(体系仅为一份样品体系)配置反转录反应液。该过程同上,试剂置于冰上或冰盒,配置反应液总管,最后向已去除gdna的样品中加入反转录反应液。将反应液分别加入管中,混匀,按表9程序进
行pcr反应。
[0185]
表8反转录体系
[0186]
4.5
×
primescript buffer 2(for real time)4μl5.rt primer mix1μl6.rnase free dh2o4μl3.primescript rt enzyme mix i1μl
[0187]
表9反转录程序
[0188]
37℃15min85℃5sec4℃保存
[0189]
反应完成,即可得到共20μl的cdna,也可根据后续试验需求对cdna进行稀释。cdna样品可于-20℃长久保存。
[0190]
9实时荧光定量pcr(qpcr)
[0191]
实时荧光定量pcr(qpcr)步骤:
[0192]
按表10配制pcr反应液(反应液的配置在冰上进行)。考虑到吸取误差,配置的预混液体积要至少多于所有反应用总体积的10%。qrt-pcr引物见表11。
[0193]
表10 pcr反应液配方
[0194][0195]
表11 qrt-pcr引物
[0196][0197][0198]
10基因组dna的提取
[0199]
基因组dna的提取步骤:
[0200]
(1)消化细胞及细胞计数:细胞消化离心后进行细胞计数,使细胞数不超过5
×
107个细胞。计数完成后,300g离心5min,弃掉上清,小心不要弃掉细胞沉淀;
[0201]
(2)向样品中加入200μl pbs重悬细胞,向样品中加入20μl qiagen protease或者
蛋白酶k;
[0202]
(3)向样品中加入200μl缓冲液al,涡旋混匀15s,为了确保裂解的高效,必须彻底混合样品和缓冲al以产生均质溶液,如果样品体积大于200μl,则等比例增加qiagenprotease(或蛋白酶k)和缓冲al;例如,400μl样品将需要40μlqiagenprotease(或蛋白酶k)和400μl缓冲液al。如果需要大于400μl的样品量,则可使用qiaampdnabloodmidi或者maxikits;这些可以处理多达2ml或10ml的样本;
[0203]
(4)56℃水浴孵育10min,在56℃下孵育10分钟,dna的产量可达到最大值。但注意,更长的孵化时间对纯化dna的产量或质量则无任何影响;
[0204]
(5)将离心管短暂离心,使管盖和管壁的液体滑落至底部;
[0205]
(6)向样品中加入200μl的乙醇(96vt%),涡旋混匀15s;混匀后,短暂离心以使管盖和管壁的液体滑落至底部,若样品体积大于200μl,则按比例增加乙醇的量;例如,400μl样品需要400μl的乙醇;
[0206]
(7)将(6)得到的混合液转移至qiaampminispincolumn(2ml收集管)中,6000g离心1min。之后将qiaampminispincolumn转移至新的2ml收集管中,弃掉旧的收集管,盖紧每个过滤柱的盖子,避免在离心过程中形成气溶胶;
[0207]
(8)打开qiaampminispincolumn并向其中加入500μl的aw1缓冲液,6000g离心1min。将qiaampminispincolumn转移至新的2ml收集管(试剂盒提供)中,并弃掉旧的收集管;
[0208]
(9)打开qiaampminispincolumn并向其中加入500μl的aw2缓冲液,盖紧盖子,全速14000rpm离心3min;
[0209]
(10)将qiaampminispincolumn转移至新的2ml收集管(试剂盒未提供)中,弃掉旧的收集管,高速离心1min;
[0210]
(11)将qiaampminispincolumn转移至新的1.5ml收集管(试剂盒未提供)中,弃掉旧的收集管。小心地打开qiaampminispincolumn盖子,并向其中加入200μl的缓冲液ae或者ddh2o。
[0211]
11蛋白质提取与免疫印迹
[0212]
蛋白质提取与免疫印迹步骤:
[0213]
11.1蛋白提取:
[0214]
(1)取出细胞,pbs洗涤细胞3次;
[0215]
(2)用细胞刮把细胞刮下至1.5ml离心管中;
[0216]
(3)放置于冰上使用裂解液裂解细胞30min;
[0217]
(4)4℃离心机12000rpm离心20min,留上清,舍弃沉淀;
[0218]
(5)bca法测定蛋白浓度;
[0219]
(6)根据蛋白体积,向细胞裂解液中加入5xsdsloadingbuffer;
[0220]
(7)100℃煮蛋白样品10分钟。
[0221]
11.2免疫印迹
[0222]
(1)制胶:上层5%(g/100ml)浓缩胶,下层根据蛋白大小选择15%(g/100ml)分离胶;
[0223]
表1215%分离胶分离胶配方(10ml)
[0224][0225][0226]
表13 5%sds-page浓缩胶配方
[0227][0228]
(2)跑胶:上样30μg,根据蛋白丰度调整上样量,恒压80v跑过浓缩胶,转120v跑到分离胶底部或者25毫安/块胶恒流待溴酚蓝触底后停止;
[0229]
(3)转膜:恒流200毫安,转膜时间依据目的蛋白大小确定,按1min转1kd计;
[0230]
(4)封闭:用含5vt%脱脂牛奶或bsa配成的封闭液在室温(25℃)封闭1小时,或是在4℃封闭 16小时(视抗体特性的不同来决定封闭的时间与温度);
[0231]
(5)孵育一抗:一抗4℃过夜,一抗浓度1:1000,根据说明书和实验结果调整;
[0232]
(6)洗膜:3*10min tbst洗脱,吐温浓度0.1%加入tbs(tbs配制见表14)中;
[0233]
(7)国产二抗1:2000倍稀释于tbst中,二抗室温孵育1小时(抗体见表15);
[0234]
(8)洗膜:3*10min tbst洗脱;
[0235]
(9)曝光:将膜泡在发光液中(发光液由a液与b液分别等比例加入),控干后放入机器,自动曝光,随后根据结果手动调整曝光时间。
[0236]
tbs配制(缓冲液,1l)
[0237]
10
×
tbs:20mm tris-hcl,ph 7.5
[0238]
表14 tbs配制
[0239][0240]
表15基因抗体
[0241][0242][0243]
12rna pμll down
[0244]
12.1rna的准备
[0245]
(1)准备质粒:获得sense、anti-sense,并克隆到pcdna3.1载体上(通过pcr获得lcdr的全长序列简称sense链,以及lcdr的反向互补链简称antisense链,并将上述序列通过酶切位点ecor i和xho i构建至载体pcdna3.1(+)),提取质粒;
[0246]
(2)质粒线性化:确认sense、antisense的3’末端序列包含的特异酶切位点,利用相应内切酶将质粒线性化;
[0247]
(3)线性化片段纯化,对于只有特异条带的反应(pcr或酶切等)产物,可以直接回收;而对于存在非特异条带的产物,则需要切下特异条带位置的凝胶后进行回收;
[0248]
注意事项:对于pcr反应产物回收,只有步骤2和3有差异。即可以直接加入等体积的pc溶液,混匀后加入到吸附柱cb2中进行回收。
[0249]
(4)体外rna转录(thermokit+thermo am8452),rna转录体系如下:
[0250]
2μl atp
[0251]
2μl ctp
[0252]
2μl gtp
[0253]
1.3μl utp
[0254]
5μl biotin utp
[0255]
2μl 10x reaction buffer
[0256]
1μg线性化dna(即至少270ng/μl)
[0257]
2μlenzymemix
[0258]
nuclease-freeh2o补齐到20μl
[0259]
37℃反应4小时;
[0260]
(5)转录产物加1μlturbodnaase(去除模板)。混匀,37℃放置15min;
[0261]
(6)上述产物中加入30μlnuclease-freeh2o和30μllicl,混匀,-20℃放置30min以上。4℃离心机12000rpm离心15min,吸弃上清;加入1ml70vt%无酶乙醇吹起,再离心,然后小心弃净上清,晾干后加适量无酶水溶解。分装后,液氮速冻,存于负八十冰箱。
[0262]
1.2celllyaste的准备
[0263]
准备6个10cm皿的细胞,密度95%。于冰上,冷的pbs(4℃)洗两次,弃净上清;每皿加入120μliplysis(加入1%inhibitor),冰上裂解5min;然后用细胞刮(提前用rnase清除剂处理)刮下,收集至离心管中,冰上放置15min;4℃离心机12000rpm离心20min;上清转移至新管中,测浓度后分装至ep管中,调整浓度,使每管含有2mg裂解液(体积1ml)。
[0264]
1.3rnapμlldown
[0265]
(1)rna形成二级结构:各取5pmol生物素标记的rna,加入50μlrnastructurebuffer(10mmtrisph7.0、0.1mkcl、10mmmgcl2),95℃放置2min,冰上放置3min,然后室温(25℃)静置30nmin以形成二级结构;
[0266]
(2)rna与蛋白结合:步骤二中细胞裂解液,从冰箱取出后,于冰上溶解,均分至两个无酶的ep管中(每管含有1mg蛋白),分别加入等量的上述形成反应液,垂直混悬仪器室温旋转1h;
[0267]
(3)各取50μlstreptavidinagrosebeads(life,m280),分别用iplysisbuffer洗5次,每次5min;将洗好的beads分别加入上述反应液中,垂直混悬仪器室温旋转1h;
[0268]
(4)用iplysisbuffer清洗beads5次,每次5min;各加入40μl1xloadingbuffer95℃煮5min;
[0269]
(5)得到样品后进行电泳,并进行相应的染色。
[0270]
13rna免疫共沉淀(rip)
[0271]
(1)细胞裂解:准备3盘10cm皿且密度为80%的nci-h1299细胞,吸弃培养基,用预冷的培养基(4℃)洗涤3次,用细胞刮将细胞刮取并放置于15ml的离心管,4℃离心机1500rpm离心5min,收集细胞。pbs重悬细胞沉淀,4℃离心机1500rpm离心5min,重复两次。向上述细胞沉淀加入200μl细胞裂解液,并用移液枪上下吹打混匀,冰上孵育10min;
[0272]
(2)清洗磁珠:使用前重悬磁珠,吸取50μl的磁珠置于1.5mlep管中,加入500ripwashbuffer,使用移液枪吹打混匀15次,置于磁力架,吸弃上清,重复清洗两次;
[0273]
(3)磁珠与抗体结合:向清洗后的磁珠中加入100μl的ripwashbuffer重悬磁珠,并加入相应的抗体5μg,室温置于旋转仪上30min,使抗体与磁珠结合;
[0274]
(4)磁珠与抗体孵育完成后,ripwashbuffer反复清洗磁珠3次,去除非特异性结合的蛋白;
[0275]
(5)磁珠-抗体复合物与细胞裂解液孵育:按表16配置结合缓冲液;细胞裂解液4℃离心机1500rpm离心20min,离心后吸取100μl细胞裂解液与900μl的结合缓冲液4℃垂直旋
转仪过夜孵育;
[0276]
表16结合缓冲液配方
[0277]
试剂名称用量(μl)rip wash buffer8600.5m edta35rnase inhibitor5total900
[0278]
(6)将上述ep管顺离,置于磁力架上,吸弃上清,加入500μl rip wash buffer并使用移液枪上下混匀20次,重复洗涤6次,去除非特异性结合的rna;
[0279]
(7)蛋白酶消化释放rna:按表17配置蛋白消化体系;将上述洗涤干净的磁珠进行蛋白消化,置于60℃振动仪上消化30min;
[0280]
表17蛋白消化体系配方
[0281]
试剂名称用量(μl)rip wash buffer11710%sds15proteinase k18total150
[0282]
(8)将上述ep管顺离,置于磁力架上,吸取上清进行rna的纯化回收。
[0283]
14染色质免疫共沉淀(chip)
[0284]
使用magna chip hisens chromatin ip kit(merck millipore)的试剂盒进行chip实验,具体操作步骤如下:
[0285]
①
提前将nci-h1299种植与6cm皿,细胞密度为90%;
[0286]
②
吸弃培养基,用预冷的pbs(4℃)清洗3次;
[0287]
③
吸弃培养基,加入fixing buffer a,同时加入11.1%(g/100ml)的新鲜的甲醛,使甲醛的终浓度为1%(g/100ml),摇床上摇动5min;
[0288]
④
吸弃上清,使用预冷的pbs(4℃)洗涤3次,除去残余的甲醛;
[0289]
⑤
细胞刮细胞刮取,并放置于1.5ml的ep管中,4℃离心机200g离心5min;
[0290]
⑥
吸弃上清,加入pbs上下混匀10次,4℃离心机200g离心5min,清洗两次;
[0291]
⑦
吸弃pbs,向上述细胞沉淀加入细胞裂解液,4℃孵育10min;
[0292]
⑧
4℃离心机1700g离心10min,吸弃上清,收集完整的细胞核;
[0293]
⑨
向上述的细胞核中加入200μl的wash buffer,使用移液枪混匀,置于4℃孵育10min;
[0294]
⑩
4℃离心机1700g离心5min,收集细胞核,吸弃上清;
[0295]
沿着ep的管壁缓慢的加入shareing buffer d3,4℃离心机200g离心5min,重复两次;
[0296]
吸弃上清,加入130μl的shareing buffer d3重悬细胞核,进行超声;
[0297]
使用移液枪混匀磁珠,并吸取10μl的磁珠置于1.5ml的ep管中,置于磁力加上,吸弃上清,同时加入50μl scw buffer蜗旋10.s,重复清洗3次;
[0298]
置于磁力加上,吸弃上清,加入200μl的scw buffer,并加入相应的抗体5μg,室温
垂直旋转仪孵育2h;
[0299]
将上述的ep管顺离,置于磁力架上,吸弃上清;
[0300]
使用移液枪加入500μl含有蛋白酶抑制剂的scw buffer,并加入10μl染色质混匀,置于4℃垂直旋转仪过夜孵育;
[0301]
将上述过夜孵育的ep管顺离,吸弃上清,并用含有蛋白酶抑制的sce buffer清洗三次;
[0302]
向ep管中加入500μl预冷的含有蛋白酶抑制剂的low stringency ip wash buffer(4℃),涡旋10s,置于磁力架,吸弃上清;
[0303]
洗脱dna:向上述的ep管中加入50μlchip elution buffer和1μl的蛋白酶k,75℃孵育15min,消化蛋白,冷却至室温,短暂离心,吸弃上清,纯化回收dna。
[0304]
15蛋白质凝胶迁移阻滞实验(electrophoretic mobility shift assay emsa)
[0305]
(1)配置5%天然非变性琼脂糖凝胶,将其置于0.5x tbe溶液中,100v预跑胶30-60min;
[0306]
(2)在预跑胶的过程中,按图2进行反应体系的配置;
[0307]
(3)将探针与纯化的hnrnp k蛋白按照上述反应体系进行孵育,室温(25℃)30min,加入5μl的loading buffer混匀;
[0308]
(4)吸取反映的样品20μl加入到琼脂糖凝胶中进行电泳,直至溴酚蓝迁移至凝胶底部四分之一为止;
[0309]
(5)转膜:剪取相应的尼龙膜浸泡于0.5tbe中5min,使用400ma的电流转膜30min;
[0310]
(6)待转膜完成,量取相应的尼龙膜的大小,并进行紫外交联;
[0311]
(7)将紫外交联后的膜置于nucleic acid detection blocking buffer中15min,并摇晃;
[0312]
(8)将封闭后的尼龙膜至于conjugate/blocking buffer中15min,并摇晃;
[0313]
(9)转移尼龙膜至wash solution buffer中5min,重复洗涤4次;
[0314]
(10)将洗涤后的尼龙膜放置于含有30ml的substrate equilibration buffer中,摇晃并孵育5min;
[0315]
(11)使用镊子夹起尼龙膜,并将其置于干燥纸上,然后将其放置于substrate working solution室温孵育5min;
[0316]
(12)进行曝光,并采集图像。
[0317]
16northern blot
[0318]
northern blot步骤:
[0319]
①
探针制备:体外转录并标记lcdr的探针(探针的核苷酸序列如seq id no:42所示);
[0320]
②
将实验中所使用的的所有仪器进行depc的处理,并进行depc水的洗涤;
[0321]
③
配置50ml 1
×
gel prep/runningbuffer,称取0.5g琼脂糖(agarose-le),并进行琼脂糖凝胶的配置;
[0322]
④
吸取准备好的25μg的总rna,加入等体积glyoxal load dye试剂,置于50℃的烘箱中孵育30min;
[0323]
⑤
将
③
中配置的凝胶置于1
×
gel prep/runningbuffer中浸泡5min,将相应的rna
加入到琼脂糖凝胶的孔中,在5v/cm的电压下进行跑胶,直至溴酚蓝转移至凝胶的底部;
[0324]
⑥
将凝胶置于虹吸装置中,转膜时间2h;
[0325]
⑦
量取尼龙膜的大小,根据大小进行相应的交联;
[0326]
⑧
提前将试剂μltrahyb置于68℃进行预热,将交联完成的尼龙膜置于10cm皿中,并加入预热的μltrahyb使其覆盖膜的表面,68℃进行预杂交30分钟;
[0327]
⑨
向
⑧
中的尼龙膜上加入终浓度0.2nm的lcdr探针,并加入相应的rna酶抑制剂,4℃杂交过夜孵育;
[0328]
⑩
将过夜孵育的尼龙膜置于3mllowstringencywashingsolution进行洗涤,并重复两次,每次10min;随后将尼龙膜置于3mlhighstringencywashingsolution溶液中,68℃洗涤两次每次20min;
[0329]
进行曝光,并采集图像。
[0330]
17细胞凋亡流式检测
[0331]
细胞凋亡流式检测步骤:
[0332]
(1)制备阳性细胞:选择1盘10cm皿细胞,打开培养皿盖子,放置于紫外灯下照射2小时。或使用碧云天细胞凋亡阳性对照试剂盒提前处理阳性细胞;
[0333]
(2)收集细胞:将待测细胞(包括阴性与阳性对照细胞)的上清分别收集至15mlep中,使用2mlpbs冲洗细胞,加入1ml不含有edta的胰酶,至37℃培养箱中消化,至细胞大片脱离皿底。用刚才收集的上清液终止消化后,转移至刚才的15mlep管中。2000rpm,室温,离心5min,弃上清;
[0334]
(3)清洗细胞:用1ml常温pbs重悬细胞,转移至1.5mlep管中。2000rpm,室温,离心5min,弃上清;
[0335]
(4)细胞计数:使用细胞计数仪进行细胞计数,每组取2
×
106细胞;
[0336]
(5)染色:使用200μl缓冲液重悬细胞(2
×
106),所有实验组与阴性对照需要双染,阳性对照需要设置两个染料的单染管。双染管加入5μlannexinv染料,5μl7-aad,单染管加入单独的5μlannexinv染料或5μl7-aad,标清楚单染染料名称。用手弹匀,避光染色15min。染色过程中准备流式管和滤膜。
[0337]
打开流式细胞仪,检查鞘液和废液情况;
[0338]
(6)染色完毕后,加入1mlpbs,2000rpm,室温,离心5min,弃上清。再加入100μlpbs重悬,过滤网;
[0339]
(7)上机设置:打开ssc,fsc,bb515下的fitc通道,以及7-aad通道,其他用不到的通道删除。使用log模式,全选a,h,w指标。设置初始读值:癌细胞ssc设置200,fsc设置200,fitc电压值设为280,7-aad设置350;
[0340]
(8)电压及补偿调节:看各自通道的峰图调节电压,使阴性峰的峰尾在102和103之间,峰靠右则降低电压,峰靠左则升高电压。上annexinv单染管时,调节7aad-fitc补偿,输入x%,使十字门一二象限没有细胞信号。上7aad单染管时,调fitc-7aad补偿,输入x%,使十字门一四象限没有细胞信号。若实验时间有限,来不及调节好补偿。fitc-7aad和7aad-fitc都输入1%,以保留补偿门,可在flowj软件上再次调节;
[0341]
(9)电压和补偿调节好后,不可再次改变。若电压发生改变,补偿应该再次调节,每管细胞收30000~50000个;
[0342]
(10)按照清洗流程标准清洗流式细胞仪,拷贝数据;
[0343]
(11)flowj调节补偿,打开flowj的界面,把两个单染管拖入补偿组。自动调节补偿后应用至所有实验组;
[0344]
根据阴性对照设置十字门,保证一二四三个象限中细胞数小于0.4%。应用阴性对照圈门至所有实验组。
[0345]
输出各组十字门结果至layouts,合理排列后,调节字体大小与格式,tiff格式输出。
[0346]
18数字pcr
[0347]
数字pcr步骤:
[0348]
(1)建立标准曲线:使用pcdna3.1(+)载体构建lcdr重组质粒,转入大肠杆菌dh-5a扩大培养后,提取重组质粒,然后使用nanodorp 2000检测质粒浓度。接着通过经验公式将质粒浓度换算为质粒拷贝数:拷贝数(copies/μl)=[质粒浓度(ng/μl)
×
1μl
×
6.02
×
1023
×
10-9
]/[(载体长度+目的基因长度)
ꢀ×2×
324.5];
[0349]
(2)按照10倍梯度稀释法将质粒连续稀释7个梯度,通过荧光定量pcr检测每个梯度样品的ct值 (各三个技术重复),然后建立log10(拷贝数)与ct值的线性关系即为标准曲线;
[0350]
(3)单个细胞拷贝数绝对定量:细胞计数后,取100万细胞进行rna提取,使用100μl depc水溶解rna,接着取1μl rna在20μl体系里逆转录为cdna,稀释到40μl后,取1μl作为模板进行荧光定量pcr得到样品ct值,然后根据标准曲线计算出目的基因在1μl模板里的拷贝数,最后除以细胞量即得到单个细胞拷贝数。
[0351]
19双荧光素酶报告实验
[0352]
双荧光素酶报告实验步骤:
[0353]
(1)使用in vitro dna&sirna transfection reagent试剂对质粒进行转染。按照试剂说明书,选择合适的转染体系,本次实验以24孔板为例,具体体系见表18;按照上表配制好转染体系后,涡旋10s,短暂离心后静置10min;
[0354]
表18转染体系
[0355]
试剂用量jetprime buffer50μl质粒dna总量500ngjetprime1μl
[0356]
(2)细胞传代,取生长状态良好的293t细胞,小心吸弃培养液上清,1xpbs洗两遍后,用胰酶消化2min,加入2ml的完全培养基终止反应,1000rpm/5min。用1ml的完全培养基轻轻重悬;
[0357]
(3)在24孔板中,每个孔内加入450μl新鲜的dmem完全培养基,并加入适量的细胞悬液,使其第二天的密度能达到60%左右,再向每个孔中加入配好的反应液,轻轻摇晃培养皿,使液体混匀,并放入细胞培养箱中培养72h;
[0358]
(4)使用dual-reporter assay system试剂盒进行双荧光素酶报告基因检测,弃上清,使用pbs洗涤两遍;
[0359]
(5)每个孔内分别加入100μl plb裂解液,于室温置摇床120rpm 10min进行裂解;
使用移液器将孔内的液体吹打均匀后,分别吸取20μl至白色全不透光的96孔酶标板中;
[0360]
(6)向板中的每孔分别加入80μl提前用pbs稀释4倍体积后的larii,置于酶标仪中,全波长检测萤火虫荧光素酶活性;
[0361]
(7)待检测结束,立即加入80μl1
×
stop&gloreagent,用移液器吹打混匀后,置于酶标仪中,全波长检测海肾荧光素酶活性;
[0362]
(8)将海肾荧光素酶活性读值作为内参归一,萤火虫荧光素酶活性比海肾荧光素酶活性得到每个组的荧光素酶活性;
[0363]
(9)将对照组的荧光素酶活性归一,分别求得各个实验组的相对荧光素酶活性。并进行统计。
[0364]
20荧光原位杂交(fish)和免疫荧光
[0365]
20.1荧光原位杂交
[0366]
(1)构建lcdr特异性探针:将与lcdr特异性结合的100bp的探针(核苷酸序列如seqidno:42所示)构建至pcdna3.1(+),将上述质粒进行单酶切,利用pcdna3.1(+)的t7启动子进行体外转录,并进行地高辛标记;
[0367]
(2)提前将目的细胞均匀的铺至共聚焦小皿中,第二天密度为60%即可;
[0368]
(3)去除培养基,使用depc水配置的pbs轻柔的清洗共聚焦小皿3次,避免吹起细胞,加入4%的多聚甲醛室温固定20min;
[0369]
(4)吸弃甲醛,加入depc水配置的pbs置于摇床洗涤3次,每次10min;
[0370]
(5)吸弃培养基,室温稍晾干共聚焦小皿,随后加入1:9稀释的胰酶覆盖共聚焦小皿的底部进行消化;
[0371]
(6)吸弃胰酶,加入depc水配置的pbs置于摇床洗涤3次,每次5min;
[0372]
(7)加入0.5%(g/100ml)triton-100(含有1%rna酶抑制剂,g/100ml),冰上放置10min,吸弃triton-100,加入depc水配置的pbs置于摇床洗涤3次,每次5min;
[0373]
(8)吸弃pbs,室温下稍晾干,加入预杂交液覆盖共聚焦小皿的底部置于52℃很恒温箱中2h;
[0374]
(9)向上述的预杂交液中加入100pmol的lcdr的特异性探针,杂交18h;
[0375]
(10)待杂交完成,吸弃预杂交液,加入提前52℃预热的2
×
ssc,洗涤3次,每次5min;
[0376]
(11)4
×
ssc中加入等体积的去离子甲酰胺,52℃预热,涤3次,每次15min;
[0377]
(12)加入1%的pbst洗涤5次,每次5min;
[0378]
(13)吸弃pbst,加入10%的山羊血清室温封闭1h,加入山羊血清稀释500倍的alexafluor647-conjugatediggfractionmonoclonalmouseanti-digoxinantibody二抗,置于4℃的冰箱过夜孵育;
[0379]
(14)吸弃二抗,加入depc水配置的pbs置于摇床洗涤3次,每次5min;
[0380]
(15)加入depc水配置的pbs的4’,6-diamidino-2-phenylindole(dapi)覆盖共聚焦小皿的底部,室温放置5min,吸弃dapi,洗涤3次,每次5min;
[0381]
(16)使用共聚焦显微镜进行观察,获取相应的结果。
[0382]
20.2免疫荧光
[0383]
(1)pbs浸洗细胞爬片3次,每次5min;
[0384]
(2)吸去pbs,4%多聚甲醛室温(或使用冷甲醇-20℃或冷丙酮,冷甲醇和冷丙酮同时带通透作用) 固定15min;
[0385]
(3)吸去固定剂,pbs浸洗3次,每次5min;
[0386]
(4)吸去pbs,0.5%triton x-100(pbs配制)室温通透20min;
[0387]
(5)吸去通透剂,pbs浸洗3次,每次5min;
[0388]
(6)吸去pbs,在玻片上滴加封闭缓冲液,室温封闭30min;
[0389]
(7)封闭结束前,按说明书的指示用抗体稀释缓冲液稀释一抗;
[0390]
(8)吸去封闭液,不洗,每张玻片滴加足量的稀释好的一抗,放入湿盒中4℃孵育过夜;
[0391]
(9)从4℃取出湿盒,室温复温15min;
[0392]
(10)吸去一抗,pbst(pbs,含1
‰
tween20)浸洗3次,每次5min;
[0393]
(11)吸水纸吸干爬片上多余液体,滴加稀释好的荧光二抗,湿盒中37℃孵育1h;
[0394]
(12)pbst浸洗3次,每次5min;
[0395]
(13)复染核:滴加dapi避光孵育5min,对标本进行染核;
[0396]
(14)pbst浸洗3次,每次5min;
[0397]
(15)载玻片上滴一小滴含抗荧光淬灭剂的封片液,将爬片从孔中取出,吸水纸吸干多余液体,面朝下盖到封片液上,使其接触到载玻片上;
[0398]
(16)将玻片置于暗处5min使其晾干,在荧光显微镜下观察并采集图像。
[0399]
21免疫组化(ihc)
[0400]
(1)芯片购买于上海芯超,芯片置于65℃烘箱中处理1h左右,观察到芯片上固定的蜡开始脱落即可,芯片相应信息见表19;
[0401]
表19芯片信息
[0402]
[0403][0404]
(2)将上述芯片完全浸泡于二甲苯中10min,重复浸泡三次左右,直至所有的蜡全部脱落;
[0405]
(3)将脱蜡后的芯片进行复水:将上述芯片依次浸泡于100vt%乙醇、95vt%乙醇、75vt%乙醇室温 (25℃)10min;
[0406]
(4)浸泡于pbs中5min,重复3次;
[0407]
(5)抗原修复:配置500ml柠檬酸钠修复液,将上述芯片注入处于沸腾状态的柠檬酸钠修复液中 30min;
[0408]
(6)待煮沸完成,将其置于室温自然冷却3h;
[0409]
(7)浸泡于pbs中5min,重复3次;
[0410]
(8)使用免疫组化笔圈出组织的位置,滴加3%过氧化氢(hydrogen peroxide,h2o2)封闭内源性过氧化物酶,于室温放置10min,pbs洗3次,每次5min;
[0411]
(9)封闭:加入10vt%的山羊血清覆盖组织的表面,置于4℃冰箱过夜;
[0412]
(10)吸弃封闭液,pbs洗涤5min,加入1:200稀释的hnrnp k以及laptm5的一抗,置于4℃冰箱过夜;
[0413]
(11)吸弃一抗,室温复温5-10min,pbs洗3次,每次5min,加入相应的二抗,室温45min,pbs 洗3次,每次5min;
[0414]
(12)配置dab显色剂,滴加于芯片,观察芯片颜色变化;
[0415]
(13)待显色完成,将芯片置于pbs中,终止显色;
[0416]
(14)加入苏木素进行复染,时间约为1min,置于pbs终止染色,加入氨水进行反蓝,室温晾干;
[0417]
(15)中性树胶封片,在荧显微镜下观察并采集图像。
[0418]
22统计分析
[0419]
每组数据进行三次独立实验重复,数据以mean
±
标准差(standard deviation,sd)或标准误(standarderror of mean,sem)
±
平均值(mean)表示。对照组与实验组间的比较采用mann-whitney检验或student’st;采用spearman法进行两个分子表达水平的相关性分析。实验数据采用graphpad或r语言软件进行分析,p《0.05表示差异具有统计学意义,图中当组间差异不具有统计学意义时,使用ns(no significant) 表示,*表示p《0.05,**p《0.01,***p《0.001。
[0420]
(二)实验结果
[0421]
1组蛋白乙酰化调控的基因表达谱
[0422]
为了研究组蛋白乙酰化调控的基因表达谱,利用曲古菌素(trichostatin a,tsa)和二甲基亚砜 (dimethyl sμlfoxide,dmso)分别处理beas-2b以及nci-h1299细胞(将tsa和dmso分别加入到beas-2b以及nci-h1299细胞的培养基中,终浓度为300nm,37℃、5vt%co2培养箱孵育24h),随后将处理后的细胞消化,加入trizol提取rna,并进行转录组测序。针对tsa处理的beas-2b以及 nci-h1299测序数据,通过设定阈值:p《0.05且|log2fc|≥0.58,分别筛选出10971个和5107个差异表达的基因(图3a-3b)。随后通过京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes, kegg)对beas-2b以及nci-h1299的差异基因进行通路富集(图3c-3d),同时将beas-2b与nci-h1299 差异表达的基因进行交集,结果显示交集的差异表达的基因为3095个(图3e),其中共同上调的基因有 1877个,共同下调的基因有745个,占所有差异表达基因的23.8%,kegg的数据库分析显示,这些交集的差异表达的基因在“转录异常调控”途径中得到显著富集,这一结果恰好支持组蛋白乙酰化在基因表达转录调控中的作用(图3f)。
[0423]
2组蛋白乙酰化调控以及癌变过程差异表达的lncrnas的筛选
[0424]
为了鉴定出组蛋白乙酰化调控,同时在癌变的过程中癌与癌旁差异表达的lncrnas,将上述tsa处理后差异表达的lncrnas和tcga数据库下载的肺癌、肾透明细胞癌、胆管癌、肝癌、胃癌、头颈腺鳞状细胞癌差异表达lncrnas做了一个系统的分析(寻找共同上下调的lncrnas),筛选出24个差异表达且同向变化的lncrnas(图4a),众所周知,h4k16ac和h3k27ac是调控基因转录的重要因子,但实验发现tsa抑制剂仅能够增加h3k27ac在肺细胞系中的表达,不能增加h4k16ac的表达(图4b),因此将上述差异表达且同向变化的lncrnas和nci-h1299 h3k27ac chip seq数据进一步整合(将上述差异表达且同向变化的lncrnas与nci-h1299 h3k27ac chip seq富集的基因进行交集)(图4c),结果显示仅有7个lncrnas可以被h3k27ac显著富集(图4d),通过对这些基因进行进一步的分析,其中 matn1-as1、snhg12、lucat1、nkila、mnx1-as1的功能已经得到了注释,最终聚焦于lcdr在肺癌中的作用。此外,使用不同浓度的tsa(终浓度0nm和300nm)处理beas-2b、nci-h1299以及hcc827 (将tsa分别加入到beas-2b、nci-h1299以及hcc827细胞的培养基中,37℃、5vt%co2培养箱孵育 24h),结果显示tsa处理之后,lcdr转录本水平(图4e-4g)以及h3k27ac在lcdr的启动子富集水平均显著提高(图4h-4j)。
[0425]
3lcdr的分子特征
[0426]
到目前为止lcdr的生物学功能尚未得到注释,为了进一步了解lcdr的生物学功能,首先通过3
′ꢀ
race和5
′
race技术,扩增出核苷酸序列如seq id no:1所示的lcdr的全长
核酸序列(图5a),发现lcdr是一个全长2013bp且只具有一个外显子lncrna,这一长度大小进一步被northern blot实验所证实(图5b)。随后将lcdr的序列输入到网站human blat search(http://genome.ucsc.edu/cgi-bin/hgblat),发现lcdr位于20号染色体(图5c)。通过鉴定出来的全长序列设计特异性引物,通过qrt-pcr检测 lcdr在不同肺癌细胞系以及癌旁组织的相对表达量,qrt-pcr结果显示lcdr在癌组织中表达量相对较高(图5d),这一结果进一步被绝对定量pcr所证实(图5e)。
[0427]
为了了解lcdr的编码潜能,通过使用一种用来区分非编码区和蛋白质编码区的比较基因组学方法,将lcdr的序列输入网站coding potential calcμlator(http://cpc.cbi.pku.edu.cn/)进行预测,同时将已经报道的pcseat、hotair长链非编码rna作为阳性对照,将gapdh、actb编码基因作为阴性对照,发现lcdr的编码能力为-1.17724、pcseat的编码能力为-0.786637、hotai的编码能力为-0.534273、 gapdh的编码能力为2.0942、actb的编码能力为2.95081,说明lcdr基本不具备编码蛋白质的能力 (图6a)。通过encyclopedia pf dna elements(encode)数据库,获得了lcdr启动子区域h3k4me1、 h3k4me2、h3k4me3、h3k9ac、h3k9me3、h3k27ac以及h3k36me3的修饰情况(图6b)。通过ucscgenome browser数据库分析,显示lcdr在物种间的保守性相对较差(图6c)。
[0428]
lncrna的定位可能预示着其发挥相应的功能,因此进一步了解lcdr的亚细胞定位。首先根据网站的预测设计一个特异性的探针(核苷酸序列如seq id no:42所示),通过二抗显色显示出lcdr在 nci-h1299细胞系中主要位于细胞核,同时随着lcdr表达量的下降,探针的荧光信号也出现相应的减弱(图7a),随后通过核质分离实验可以看出gapdh主要位于细胞质,histone h3主要位于细胞核,同时目标基因lcdr主要位于细胞核(图7b),进一步验证了原位杂交的实验结果。
[0429]
4c-jun调控lcdr的转录
[0430]
为了进一步明确肺癌细胞中驱动lcdr表达的分子机制,通过encode数据库预测发现lcdr的启动子区域存在显著的c-jun富集(图8a)。为了验证网站预测的结果,使用慢病毒感染nci-h1299敲降 c-jun(图8b),或过表c-jun(图8c),检测lcdr表达量的变化情况。结果显示,敲降c-jun抑制了 lcdr的表达,而过表c-jun促进了lcdr的表达。接着,通过分析gse92783测序数据,预测出两个潜在的结合位点bs1:gaggcggaagtg以及bs2:ggtttccggggt(图8d)。随后通过chip实验进行验证,结果显示与对照组igg相比,c-jun可以显著富集lcdr启动子(图8e)。同时,根据网站预测的结合位点,即-454bps~-465bps与-500bps~-511bps,对这两个区域进行了插画突变(突变序列见图8f),通过双荧光素酶实验结果显示c-jun过表达能够增强含野生型和mut2 lcdr启动子区域的双荧光素酶活性,而不影响含mut1 lcdr启动子区域的双荧光素酶活性(图8g)。此外chip-qpcr的结果显示,tsa 处理之后c-jun可以更多的富集在lcdr的启动子区域(图8h),激活lcdr的转录。综上所述,这些数据表明c-jun调控lcdr的转录。
[0431]
5敲降lcdr诱导细胞死亡
[0432]
上述的实验结果揭示,lcdr在肺癌里高表达,可能是一个潜在的癌基因,为了进一步验证lcdr在肺癌中的分子功能,针对lcdr设计了特异性的shrna和sirna(引物序列见表20),同时将lcdr 构建至plvx-ires-neo载体上,qrt-pcr显示lcdr的敲除效率(图9a-9b)以及lcdr的过表倍数(图 9c)。
[0433]
敲降lcdr的表达,导致肺癌细胞系nci-h1299的增殖能力显著下调(图10a),过表达lcdr可以促进肺癌细胞系nci-h1299的增殖(图10b)。
[0434]
敲降lcdr的表达,导致肺癌细胞系nci-h1299的克隆形成能力显著下调(图11a),过表达lcdr 可以显著提升肺癌细胞系nci-h1299的克隆形成能力(图11b)。
[0435]
此外,还发现敲降lcdr细胞出现了形态变化和凋亡样特征,如细胞收缩、变圆、起泡和脱落(图12a-12b)。为了准确的评估细胞凋亡的比率,通过fitc-annexin v和7-aad进行染色,annexin v特异性的与凋亡早期外翻的磷脂结合,7-aad特异性的与凋亡晚期细胞核内核酸进行结合。流式细胞术结果显示:与对照组相比,敲减lcdr细胞凋亡的比例显著增加。
[0436]
为了进一步说明lcdr对细胞凋亡的影响,通过免疫荧光检测了cytochrome c的亚细胞定位,结果显示慢病毒包装shrna敲降lcdr,cytochrome c从线粒体中释放至整个细胞质(图13a-13b),随后又检测了凋亡关键蛋白caspase3以及parp的表达情况,western blot结果显示,敲降lcdr、cleavedcaspase3以及cleaved parp均被检测到(图13c-13d)。综上所述,这些数据表明,敲降lcdr导致肺癌细胞凋亡。
[0437]
为了进一步研究lcdr的表达量对肺腺癌细胞肿瘤形成能力的影响,通过shrna系统敲降lcdr,获得低表达lcdr的nci-h1299细胞株,将上述对照组以及稳定低表达lcdr的nci-h1299细胞株以相同数量的细胞注射入小鼠皮下(注射在腋下部位),结果显示降低lcdr显著降低了肺腺癌细胞nci-h1299 的肿瘤形成能力(图14a-14c)。同时,将从小鼠腋下取出的肿瘤进行切片,包埋,免疫组化实验结果显示:与对照组相比敲降lcdr,cleaved caspase3的蛋白水平显著上调(图14d-14e)。总的来说,这些数据表明lcdr促进了肺癌细胞的存活。
[0438]
表20 qrt-pcr引物
[0439][0440]
6lcdr与hnrnp k特异性结合
[0441]
大量的文献表明lncrna本身是基本不具备功能,它通过形成二级结构,与蛋白质、dna以及rna 相互结合,从而发挥其生物学功能。为了寻找lcdr可能结合的蛋白质,将lcdr以及lcdr的反向互补链构建至pcdna3.1载体上,利用t7启动子进行体外转录,然后利用生物素进行标记。将制备好的反向互补链以3pmol的终浓度与nci-h1299蛋白质裂解液过夜孵育,将孵育好的复合物通过磁珠进行分离以及洗脱。通过蛋白质凝胶电泳和银染技术,发现lcdr的正义链60kd左右相对于反义链存在一条特异性条带(图15a)。蛋白质谱技术鉴定特异性条带的氨基酸,最终通过比对蛋白质的大小,蛋白质匹配的肽段数以及特异性肽段数
鉴定该蛋白可能为hnrnp k,见表21。对lcdr pull down样品进行westernblot验证,结果显示该条带为hnrnpk且是lcdr特异性结合蛋白分子(图15b)。为了进一步验证rnapμll down的实验结果,通过rip实验进行反向验证,结果显示hnrnp k能够特异性的结合lcdr(图15c)。lcdr原位杂交和hnrnp k免疫荧光显示lcdr和hnrnp k在细胞核内共存,进一步支持了它们之间的相互作用(图15d)。
[0442]
为了进一步鉴定hnrnp k与lcdr的结合位点,首先构建截短型的lcdr:第一段1-500bp;第二段 500-1000bp;第三段1000-1500bp;第四段1500-2013bp。将截短的lcdr构建至pcdna3.1载体,利用 t7启动子体外转录标记生物素后与nci-h1299蛋白裂解液孵育,通过western blot显示hnrnp k与1-500bp的lcdr相互结合(图16a)。为了进一步识别与hnrnp k结合的特定碱基,通过mfold和rnafold 网站对lcdr进行二级结构的预测,筛选出一个潜在的hnrnp k与lcdr相互结合的核酸序列: ccccccacc(图16b)。假设该聚(c)位点对于hnrnp k结合是必不可少的,因此将结合位点进行突变(突变位点见图16c),并通过rna pμll down实验反向验证,结果显示与对照相比突变型的lcdr 与hnrnp k的结合能力明显下调(图16d)。与rna pμll down结果一致,凝胶迁移阻滞实验 (electrophoretic mobility shift assay,emsa)显示与对照组相比突变型的lcdr探针与hnrnp k蛋白的结合能力明显下调(图16e)。最后通过rip实验进行反向验证,结果显示:与对照组相比过表lcdr 增强与hnrnp k的相互结合,突变lcdr减弱与hnrnp k的相互结合(图16f)。
[0443]
hnrnp k主要包含3个结构域,为了鉴定出lcdr结合hnrnp k的结构域,将hnrnp k蛋白进行截短(截短过程见图17a),并在截短体的末端连接标签蛋白flag,通过慢病毒包装对截短型的hnrnp k 进行表达(图17b)。通过rip实验,用flag磁珠与截短型的蛋白裂解液进行孵育,结果显示lcdr结合在hnrnp k的kh1(图17c)。综上所述,这些数据表明lcdr与hnrnp k相互结合。
[0444]
表21蛋白质谱鉴定lcdr潜在的结合蛋白
[0445][0446]
7敲降hnrnp k诱导细胞死亡
[0447]
由于lcdr与hnrnp k之间的相互作用,推测hnrnp k可能对癌细胞存活有着与lcdr一致的作用,因此设计了两段特异性的sirna(sirna序列见表22),同时将hnrnp k构建至plvx-ires-neo过表达载体上,qrt-pcr显示hnrnp k的敲降效率(图18a)以及hnrnp k的过表达倍数(图18b)。
[0448]
敲降hnrnp k导致肺癌细胞系nci-h1299的增殖能力明显下调(图19a),过表达hnrnp k可以促进肺癌细胞系nci-h1299的增殖(图19b)。
[0449]
敲降hnrnp k导致肺癌细胞系nci-h1299的克隆形成能力明显下调(图20a),过表
adhesion)、可变剪切(spliceosome)、内吞(endocytosis)、溶酶体(lysosome)等信号通路(图25c);hnrnp k的差异表达基因主要富集在嘌呤代谢(purinemetabolism)、肌动蛋白细胞骨架的调节(regμlation of actin cytoskeleton)、mapk信号通路(mapk signalingpathway)、胰岛素信号通路(insμlin signaling pathway)、溶酶体(lysosome)等信号通路(图25d)。接着,对lcdr的测序数据和hnrnp k的测序数据进行交集,发现有大量共同差异表达的基因共计4066 个(图25e-f),对这些基因进行kegg通路富集,分析发现这些基因主要富集在p53信号通路 (p53_signaling_pathway)、小细胞肺癌(small cell lung cancer)、细胞周期(cell cycle)、溶酶体(lysosome)、肌动蛋白细胞骨架(regμlation of actin cytoskeleton)等信号通路(图25g)。通过对上述的通路富集综合分析发现lcdr与hnrnpk存在着9个共同调控的信号通路,表明lcdr与hnrnp k之间存在功能的相互作用。溶酶体细胞死亡又称溶酶体依赖性细胞死亡,是溶酶体膜通透性改变后释放到胞浆中的水解酶介导的一种调节性细胞死亡。我们推测lcdr/hnrnp k可能共同调节溶酶体信号死亡通路。通过对溶酶体信号通路的进一步分析,挑选出溶酶体通路中与对照组相比差异变化最大的6个基因作为候选基因: arsb、asah1、hnsgat、laptm5、acp2、glb1(图25h-i)。为了进一步验证测序数据,敲降lcdr 以及hnrnp k,检测候选基因转录本的变化情况(图25j-k),qrt-pcr结果显示敲降lcdr以及hnrnpk上述6个候选基因有5个均可以被验证。
[0460]
值得注意的是,laptm5表达在溶酶体的膜上。与转录本水平一致,敲降lcdr、hnrnp k导致 laptm5的蛋白水平下调(图26a-b),相应的过表lcdr、hnrnp k导致laptm5的转录本(图26c-d) 以及蛋白水平上调(图26e-f)。此外,免疫荧光显示内源性以及过表达的laptm5均可以与lamp1(图 26g-h)共定位,上述结果表明laptm5表达于溶酶体膜上,并受到lcdr/hnrnpk轴的调控。
[0461]
11敲降laptm5诱导细胞的死亡
[0462]
上述结果表明,laptm5的表达受lcdr/hnrnp k轴调控,推测laptm5可能具有与lcdr/hnrnpk一致的作用,因此设计了两段特异性的sirna(sirna序列见表23),同时将laptm5构建至 plvx-ires-neo过表达载体上,qrt-pcr显示laptm5的敲降效率(图27a)以及laptm5的过表达倍数(图27b)。
[0463]
敲降laptm5导致肺癌细胞系nci-h1299的增殖能力明显下调(图28a),过表达laptm5可以促进肺癌细胞系nci-h1299的增殖(图28b)(使用靶向laptm5的sirna转染nci-1299 48h,敲降 laptm5的表达;将laptm5的全长序列构建至plvx-ires-neo,随后进行慢病毒包装以及感染,过表达laptm5)。
[0464]
敲降laptm5导致肺癌细胞系nci-h1299的克隆形成能力明显下调(图29a),过表达laptm5 可以促进肺癌细胞系nci-h1299的克隆形成能力(图29b)。
[0465]
敲降laptm5导致nci-h1299细胞在光镜下出现与lcdr以及hnrnp k相似的凋亡样形态变化(图 30a),流式细胞术结果显示敲降laptm5与对照组相比凋亡细胞的比例明显增多(图30b-c),免疫荧光结果显示敲降laptm5,cytochrome c从线粒体中释放至细胞质(图30d),western blot结果显示敲降laptm5,cleaved caspase3以及cleaved parp均被检测到(图30e)。
[0466]
表23 qrt-pcr引物
[0467][0468]
12lcdr、hnrnp k和laptm5共同维持溶酶体膜完整性
[0469]
根据lcdr/hnrnp k/laptm5轴对细胞生存有着一致的影响,因此推测lcdr/hnrnp k/laptm5 轴可能通过共同维持溶酶体膜的完整性来调节细胞生存。因此通过溶酶体染料(lyso tracker)(图31a)、溶酶体膜的关键蛋白lamp1、以及溶酶体内ctsb(图31b)免疫荧光染色对溶酶体膜的完整性进行评估,结果显示敲降lcdr、hnrnp k和laptm5均会导致lyso tracker,lamp1以及ctsb由聚集变得弥散,同时与对照组相比lamp1与ctsb的共定位出现明显的减弱;与免疫荧光结果一致,扫描电镜显示敲降lcdr、hnrnp k和laptm5溶酶体的膜完整性被破坏,出现明显的断裂(图31c)。综上所述,这些数据表明lcdr/hnrnp k/laptm5轴共同调节溶酶体膜的完整性。
[0470]
13.lcdr与hnrnp k通过laptm5调控细胞死亡
[0471]
鉴于上述发现,确定laptm5是否为lcdr/hnrnp k轴关键下游效应分子成为本研究的关键。首先进行细胞增殖以及克隆形成能力的挽救实验,结果显示敲降lcdr或hnrnp k对肺腺癌细胞增殖以及克隆形成的抑制作用,均能通过过表laptm5获得部分的挽救(图32a-d)。与细胞增殖以及克隆形成的结果一致,流式细胞术显示laptm5的过表达可以使敲降lcdr或hnrnp k所导致的细胞凋亡得到部分的挽救(图32e-h)。研究表明,组织蛋白酶在促进溶酶体细胞死亡中起主要作用,阻断组织蛋白酶活性可以抑制溶酶体细胞死亡(tang,kang,berghe,vandenabeele,&kroemer,2019)。ca-074me是一种高度选择性的ctsb(cathepsin bctsb)抑制剂(bogyo et al.,2000),因此探讨ctsb抑制剂对 lcdr/hnrnp k/laptm5耗竭后细胞死亡的阻断作用,结果显示ca-074me可以部分挽救敲降lcdr, hnrnp k以及laptm5所导致的细胞凋亡(图32i-n)。
[0472]
为了进一步研究lcdr/hnrnp k/laptm5轴对肺腺癌细胞肿瘤形成能力的影响,进行小鼠体内皮下瘤的挽救实验,结果显示敲降lcdr或hnrnp k对肺腺癌细胞肿瘤形成的抑制作用,均能通过过表 laptm5获得部分的挽救(图33a-c)。
[0473]
同时,将取出的肿瘤进行切片,包埋,免疫组化实验结果显示:与对照组相比,敲降lcdr细胞质中cleaved caspase3的表达量上调,过表laptm5可以促进cleaved caspase3的表达水平显著回降(图 34a-b)。综上所述,这些数据表明lcdr和hnrnp k通过laptm5促进细胞生存。
[0474]
14lcdr与hnrnp k共同调控laptm5的稳定性
[0475]
hnrnp k主要通过与靶rna结合发挥作用,为了确定hnrnp k是否与laptm5结合,接下来将 lcdr rnaseq、hnrnp k rna seq、hnrnp k rip-seq以及hnrnp k-eclip的基因进行交集分析,共获得398共同调控的基因(图35a),通过kegg进行通路富集显示:398个基因可以有效地富集出溶酶体(lysosome)等信号通路(图35b)。同时hnrnp k rip-seq以及hnrnp k-eclip数据分析显示出hnrnpk可以有效的富集出laptm5(图35c)为了进一步鉴定出lcdr和hnrnp k与laptm5的结合位点,通过双酶报实验将lcdr和hnrnp k分别与laptm5的编码区(coding sequence,cds)以及3’端非翻译区(3-untranslatedregion,3’utr)共转(将lcdr
以及hnrnp k分别与laptm5的编码区以及3’端非翻译区的质粒进行共转染48h,随后收取细胞,进行荧光素酶活性的检测),结果显示与3
′
utr共转,可以明显提高双酶报的活性,揭示lcdr和hnrnp k结合在laptm5的3
′
utr(图35d)。接下来对3
′
utr 碱基序列进行分析发现,在3
′
utr的91-107(图35e)碱基反复出现胞嘧啶,表明其可能是hnrnp k潜在的结合位点。将上述结合位点进行突变(结合位点以及突变见图35f),并构建至pcdna3.1(+)载体,体外转录标记并进行pμll down实验,结果显示laptm5的3
′
utr可以有效地结合hnrnp k,突变 laptm5-3
′
utr与hnrnp k的相互结合能力显著减弱(图35g)。同时将laptm5-3
′
utr以及mutlaptm5-3
′
utr探针(探针序列如seq id no:59和seq id no:60所示)与纯化的hnrnp k蛋白孵育,通过emsa实验结果显示仅有laptm5-3
′
utr可以有效地与hnrnp k蛋白质相互结合,并减缓hnrnp k 蛋白质的迁移速率(图35h)。此外通过rip实验进行反向验证,结果显示:与对照组相比过表 laptm5-3
′
utr显著增强与hnrnp k的相互结合,mut laptm5-3
′
utr与hnrnp k的结合能力显著下调(图35i)。最后还利用kh结构域的突变体探索了laptm5结合hnrnp k的结构域,rip-qpcr结果显示hnrnp k的kh3结构域可以有效地富集laptm5(图35j)。综上所述,这些数据表明laptm5 的3
‘
utr中的聚(c)位点可以直接与hnrnp k的kh3结构域相互作用。
[0476]
在nci-h1299细胞系敲降lcdr水平上过表达hnrnp k,同时在敲降hnrnp k水平上过表达lcdr,然后将细胞种植在6孔板中待贴壁后使用终浓度5μm的放线菌素d溶液(actinomycin d)处理细胞,选取3个不同的时间点(0h、12h、24h)收取细胞,进行laptm5转录水平的检测。上述的结果显示敲降lcdr以及hnrnp k显著降低laptm5的稳定性,过表lcdr以及hnrnp k显著提高laptm5的稳定性,但是敲降lcdr过表hnrnp k,以及敲降hnrnp k过表lcdr、laptm5的稳定性并没有得到显著的提升(图36a-b)。同时laptm5转录本表达量的检测也得到了一致的结果,敲降lcdr以及hnrnpk显著降低laptm5的表达量,过表lcdr以及hnrnp k显著提高laptm5的表达量,但是敲降lcdr 过表hnrnp k,以及敲降hnrnp k过表lcdr、laptm5的表达量与敲除组相比没有显著的上调(图 36c-d)。此外通过双酶报实验技术,进一步验证lcdr和hnrnp k结合在laptm5的3
′
utr(图36e-f)。综上所述,这些数据表明lcdr与hnrnp k相互结合共同调控laptm5的稳定性。
[0477]
上述研究已经表明过表lcdr或hnrnp k可以显著提高laptm5的稳定性以及表达量,接下来,通过hnrnp k调控laptm5转录本的表达。通过rip实验结果显示敲降lcdr,导致hnrnp k对于laptm5 的富集明显的减弱(图37a);使用慢病毒感染系统过表达lcdr,促进hnrnp k对于laptm5的富集 (图37b);通过rna pull down实验结果显示敲降hnrnp k,与对照组相比lcdr与laptm5的结合能力显著下调(图37c);使用慢病毒感染系统过表达hnrnp k,与对照组相比lcdr与laptm5的结合能力显著上调(图37d)。使用蛋白酶k处理lcdr的rna pull down样品后,再进行磁珠洗涤并纯化回收rna,这时lcdr对laptm5结合能力显著减弱(图37d),以上这些结果说明,lcdr依赖于 hnrnp k结合laptm5(图37e)。据报道,hnrnp k的kh结构域中的gxxg基序是与rna多聚(c) 位点相互作用的关键氨基酸序列(siomi,choi,siomi,nussbaum,&dreyfuss,1994),同时lcdr和laptm5 分别与hnrnp k的kh1和kh3结构域结合,因此构建了kh1和kh3的突变体,将kh1和kh3中的 gxxg突变为geeg(如图37f,在野生型hnrnp k质粒基础上,将kh1结构域gxxg突变为geeg,形成的质粒即为mut-kh1,将kh3结构域的gxxg突变为geeg,形成的质粒即为mut-kh3)。随后将 lcdr与hnrnp k,mut-kh1以及
mut-kh3共转染,通过rip(图37g),以及双荧光素酶报告实验(图 37h)的结果表明lcdr与hnrnp k的kh1的结合可以显著增强hnrnp k kh3与laptm5的结合。
[0478]
emsa实验显示lcdr和laptm5-3
′
utr可以分别与hnrnp k的蛋白相互结合,二者与纯化的 hnrnp k蛋白共孵育(将lcdr和laptm5-3
′
utr分别与hnrnp k的蛋白相互孵育30min,随后将孵育后的样品进行跑胶、转膜、显色),进一步加强了与hnrnp k蛋白的相互结合(图38a),提示lcdr 和laptm5 3
‘
utr可能与hnrnpk具有协同结合作用。lcdr rna探针与hnrnp k蛋白孵育,加入 laptm5-3
′
utr不能进一步加强与hnrnp k蛋白的相互结合(图38b-38c);laptm5 rna探针与hnrnpk蛋白孵育,加入lcdr加强了与hnrnp k蛋白的相互结合(图38d-38e)。综上所述,这些数据表明 lcdr促进hnrnp k结合laptm5。
[0479]
15lcdr/hnrnp k轴的表达在肺腺癌中的意义
[0480]
上述研究已经表明lcdr是一个具有重要生物学功能的lncrna,为了进一步研究lcdr的临床意义,使用82对配对样本以及16个单独肺腺癌的组织芯片(从上海芯超购买了组织芯片,共计180组织点,其中包括82对配对的癌与癌旁组织,以及只具有16个组织点的癌组织),通过原位杂交实验,得出与细胞水平以及tcga数据库一致的结果,lcdr癌组织的表达量显著高于癌旁组织(图39a-39b)。同时,基于这套组织芯片(lcdr hnrnp k以及laptm5的染色均使用这套组织芯片),免疫组化技术显示出hnrnp k(图39c-39d)以及laptm5(图39e-39f)在癌组织中的表达量也同样高于癌旁组织。
[0481]
上述实验证实了lcdr与hnrnp k相互作用调控laptm5的表达,接下来探究lcdr与hnrnp k 表达量是否存在显著的相关性。基于上述的这套组织芯片,得出每个组织点lcdr,hnrnp k以及laptm5 的相对表达量(原位杂交以及免疫组化实验之后,会对组织芯片染色强度进行评估,即会得出每个组织点lcdr hnrnpk以及laptm5的相对表达),通过线性相关分析显示出lcdr与hnrnp k以及laptm5 (图40a-b)在组织表达量存在显著的相关性。hnrnp k和laptm5表达量没有相关性图(40c),表明hnrnp k调控laptm5的复杂性。同时受试者工作曲线(receiver operator characteristic curve,roc) 联合分析显示出更大的诊断价值以及临床意义(图40d)。
[0482]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1