一种不对称催化制备替格瑞洛中间体的方法与流程
1.本发明属于医药合成技术领域,涉及替格瑞洛中间体的不对称催化制备方法,具体涉及替格瑞洛重要中间体(s)-(3,4-二氟苯基)乙二醇和中间体(s)-(3,4-二氟苯基)-环氧乙烷的合成制备方法。
背景技术:
2.替格瑞洛是一种血小板聚集抑制剂,为新型的环戊基三唑嘧啶类口服抗血小板药物。替格瑞洛为非前体药,无须经肝脏代谢激活即可直接起效,能与血管平滑肌细胞上的嘌呤2受体(purinoceptor 2,p2)亚型p2y12可逆性结合,对二磷酸腺苷引起的血小板聚集有明显的抑制作用,能有效改善急性冠心病患者的症状,降低血栓性心血管事件的发生率。
3.替格瑞洛的化学结构如下:
[0004][0005]
其中,(1r,2s)-2-(3,4-二氟苯基)环丙胺是合成替格瑞洛过程中的关键中间体,结构如下:
[0006][0007]
(1r,2s)-2-(3,4-二氟苯基)环丙胺的制备方法最早由专利wo 2008018822公开。化合物2-氯-1-(3,4-二氟苯基)乙-1-酮1,经cbs还原得到手性化合物(s)-2-氯
ꢀ‑
1-(3,4-二氟苯基)乙醇2,再经碱的作用环化得(s)-(3,4-二氟苯基)环氧乙烷3,其与磷酰基乙酸三乙酯经环丙烷化反应得化合物4,再经氨解、霍夫曼降解制得替格瑞洛关键中间体(1r,2s)-2-(3,4-二氟苯基)环丙胺6。
[0008][0009]
但该方法中cbs还原反应的催化剂性质不稳定,未商品化,需要原位制备,从而增加了操作工序,不利于工业化生产;且该反应易释放出恶臭的二甲硫醚气体,对环境不友好,更不利于生产的劳动保护。此外,该方法在cbs还原中的化合物2的光学纯较低,仅有76%ee,导致最后得到的关键中间体5的光学纯也不高,为81%ee,难以满足药物对光学纯的要求。
[0010]
此后,有一些报道针对该还原工艺进行了优化。如专利cn104974017a报道了cbs还原反应优化工艺,将ee值由76%提高至98-99%。由于该反应对反应条件的控制较为严苛,反应放大时淬灭反应放热剧烈,存在较大的安全隐患。后续也有一些专利(cn 107814692,cn 107892693,cn 108083997)对cbs还原反应及其它步骤进行了优化,但未能从根本上解决问题。
[0011]
上述的方法得到手性化合物(s)-2-氯-1-(3,4-二氟苯基)乙醇2为化学法,通过生物催化法也可以对氯代酮衍生物1进行不对称还原得到手性氯代醇2。文献 cn201610051136.4报道chkred20羰基还原酶,可立体选择性还原本发明所述底物。专利cn 106701840、wo 2018/090929、cn 107686447、cn 111073919等报道了手性氯乙醇的生物酶催化还原技术。酶催化方法有比较好的立体控制性,但有时需要在两相介质中反应,大多数情况下不能够转化完全,原料损耗较大,此外,酶的用量也很大,反应物浓度低,产生的废水量较大。
[0012]
文献cn 103073525报道了从3,4-二氟苯乙烯经sharpless不对称双羟基化生成 (s)-(3,4-二氟苯基)乙二醇iii,再通过“一锅法”与原乙酸三乙酯缩合、溴化、成环合成(s)-(3,4-二氟苯基)环氧乙烷v,合成路线如下:
[0013][0014]
文献cn 109956916报道了一种以手性salen-锰络合物作为催化剂,3,4-二氟苯乙
烯为底物,经不对称环氧化反应生成(s)-2-(3,4-二氟苯基)环氧乙烷的方法,虽然步骤较短,但其催化剂用量大,工业化上存在一定的困难。
[0015]
现有技术方案中存在立体选择性差、反应活性低、原料损耗大以及合成工艺复杂的问题。本发明中通过发展高效、绿色、高立体选择性的方法和工艺,提高替格瑞洛中间体的合成效率。
技术实现要素:
[0016]
为了克服现有技术的不足,本发明的目的在于提供一种替格瑞洛的制备方法,该方法以手性的过渡金属配体络合物为催化剂,以氢气作氢源,能够在温和的条件下实现替格瑞洛的手性制备,具有配体廉价易得,反应简洁,收率高、对映选择性好、成本低、绿色环保等优点。
[0017]
首先,本发明提供了一种下式化合物(iii)的制备方法,化合物(iii)为(s)-(3,4
‑ꢀ
二氟苯基)乙二醇,合成方法包括:
[0018][0019]
其中,式(ii)所示化合物在过渡金属催化剂的存在下,加入催化量的碱,于氢气氛和有机溶剂中反应,生成式(iii)所示化合物;
[0020]
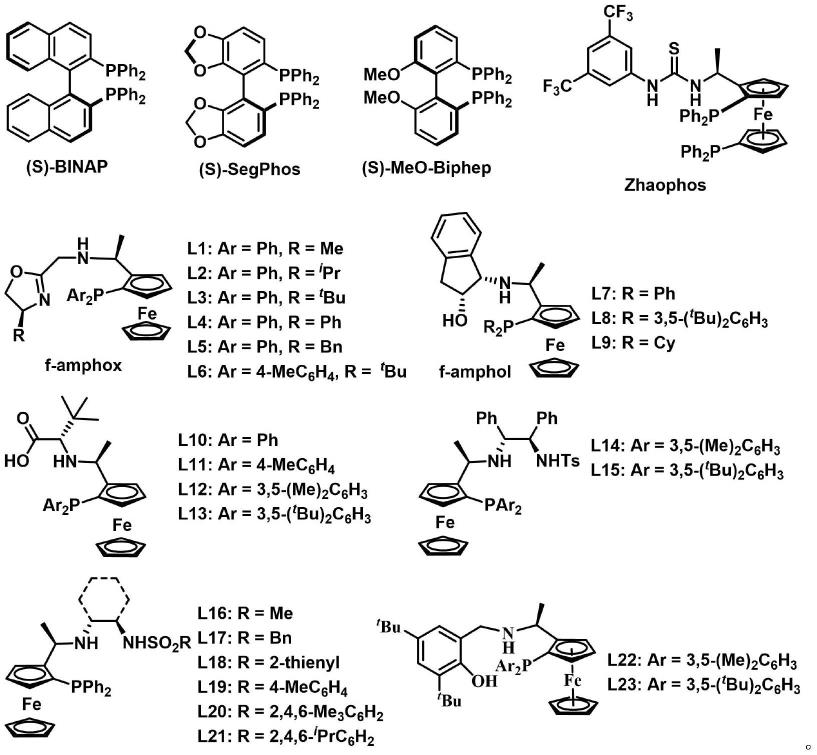
所用过渡金属催化剂由金属盐和手性配体混合后生成,催化剂金属盐选自钌、铑、铱、钯等常见过渡金属化合物,手性配体选自:
[0021][0022]
作为本发明中不对称催化步骤的一种优选技术方案,所述配体选自l3、l6 和l15,具有以下r和ar基团组合的化合物:
[0023]
l3:ar=ph,r=tbu;
[0024]
l6:ar=4-mec6h4,r=
t
bu;
[0025]
l15:ar=3,5-(
t
bu)2c6h3。
[0026]
作为本发明中不对称催化步骤的一种优选技术方案,所述过渡金属优选为铱,具体地为[ir(cod)cl]2。
[0027]
本发明提供的不对称催化氢化反应的体系为均相体系。
[0028]
作为本发明中不对称催化步骤的一种优选技术方案,所述均相催化氢化反应在含有甲醇、乙醇、异丙醇、四氢呋喃、二氯甲烷、甲苯的一种或任意比例的混合溶剂中进行,优选地为醇类溶剂。
[0029]
作为本发明中不对称催化步骤的一种优选技术方案,所用的碱为叔丁醇钾、叔丁醇钠、叔丁醇锂、氢氧化钾、氢氧化钠、碳酸钠、碳酸钾、碳酸铯的一种或任意比例的混合物。
[0030]
作为本发明中不对称催化步骤的一种优选技术方案,所述反应的温度为 20-80摄氏度,更优选30-60摄氏度;所述反应的氢气压力为1-8mpa,更优选 3-5mpa。
[0031]
作为本发明中不对称催化步骤的一种优选技术方案,所述反应时间为4-60 小时。
[0032]
作为本发明中不对称催化步骤的一种优选技术方案,所述中间体(ii)与催化剂的摩尔比为2mmol:0.01-1nmol。
[0033]
本发明还提供了一种下式化合物(v)的制备方法,合成路线包括:
[0034][0035]
具体地,合成步骤包括:
[0036]
(1)以2-氯-1-(3,4-二氟苯基)乙-1-酮(i)为原料,通过甲酸钠水解得到2-羟基-1-(3,4-二氟苯基)乙-1-酮(ii);
[0037]
(2)通过权利要求1-7任一项所述的制备方法由化合物(ii)制备得到化合物 (iii);
[0038]
(3)化合物iii在催化量的二丁基氧化锡作用下,形成伯醇的单磺酰化中间体(iv);
[0039]
(4)通过碱的作用下,由化合物(iv)分子内环化得到(s)-(3,4-二氟苯基)-环氧乙烷(v)。
[0040]
作为前述方法的一种优选技术方案,所述步骤(3)中,(s)-(3,4-二氟苯基)
‑ꢀ
环氧乙烷(v)的合成方法包括:
[0041][0042]
具体地,化合物iii在催化量的二丁基氧化锡作用下,形成伯醇的单磺酰化中间体(iv),通过碱的作用下分子内环化得到(s)-(3,4-二氟苯基)-环氧乙烷(v),其中,二丁基氧化锡的量优选为0.5-5mol%。
[0043]
本发明进一步提供了一种新颖的中间体化合物,所述化合物结构如下:
[0044][0045]
本发明进一步提供了一种不对称催化制备替格瑞洛的方法,由所述的制备方法制备得到中间体(v),进一步制备得到替格瑞洛。
[0046]
其中,由中间体(v)进一步制备得到替格瑞洛的方法可以是现有技术已知的制备方法,例如:
[0047][0048]
本发明相对于现有技术的有益效果包括:
[0049]
(1)本发明利用不对称催化氢化技术合成的中间体(s)-(3,4-二氟苯基)乙二醇(iii),操作简便,接近当量的产物转化,ee值高达99%,具有优异的收率和立体选择性控制;
[0050]
(2)另外,催化剂用量少且催化效率高,改善了反应活性,原料损耗少,使替格瑞洛的整体合成工艺操作简便、降低废弃物排放、成本大幅下降、易于产业化应用;
[0051]
(3)从实验结果发现,在不对称催化氢化步骤中,不同配体应用于本发明中间体iii的合成时,在收率和转化率表现出一定的特异性;
[0052]
(4)本发明还提供了一种制备替格瑞洛中间体(s)-(3,4-二氟苯基)环氧乙烷的方法,通过催化量的二丁基氧化锡,提高反应选择性和反应活性,高产率得到目标产物。
附图说明
[0053][0054]
图1,本发明替格瑞洛重要中间体的合成路线示意图。
[0055]
图2,文献报道制备手性中间体iv的合成路线示意图。
[0056]
图3,手性化合物iii的hplc谱图。
[0057]
图4,消旋化合物iii的hplc谱图。
[0058]
图5,化合物iv的核磁氢谱。
[0059]
图6,化合物iv的核磁碳谱。
[0060]
图7,手性化合物iv的hplc谱图。
[0061]
图8,消旋化合物iv的hplc谱图。
具体实施方式
[0062]
下面结合实施例和附图对本发明作进一步详细的描述,但发明的实施方式不限于
此。
[0063]
实施例1
[0064][0065]
将氯代酮底物(14.1ml,100mmol)溶于100mldmf中,加入水75ml,乙醇275ml,加入甲酸钠(20.4g,300mmol)加热至回流,回流5小时后,冷却到室温,减压旋走乙醇,加水稀释,用dcm萃取,有机相干燥旋干,快速过硅胶柱,粗产物用mtbe重结晶,得纯产物(ii)12.2g,产率为71%。
[0066]
实施例2
[0067][0068]
在氩气氛围下,将[ir(cod)cl]2(3.4mg,0.005mmol)和手性配体 f-amphox-t
bu-l3(5.8mg,0.0105mmol)溶于2ml异丙醇中,在室温条件下搅拌3小时,得到橙色澄清溶液。用微量注射器取该橙色溶液116ul,加入到中间体(ii)(1g,5.8mmol)、异丙醇(2ml)和碳酸钾(8mg,0.058mmol)的混合体系中。将反应体系置于高压釜中,用氢气置换高压釜中的气体三次,最后充入50atm氢气,在25℃下反应12小时。反应结束后,缓慢释放高压釜中的气体,加入50ml二氯甲烷,水洗,饱和食盐水洗,无水硫酸钠干燥,减压浓缩得到无色油状液体1g,即氢化产物(iii),产率为99%,经hplc分析,测得ee值为》99%。
[0069]
化合物iii数据表征如下:[α]
d25
=+33.7(c=1.00,chcl3).1h nmr(400mhz, cdcl3)δ7.18
–
7.08(m,2h),7.02
–
6.99(m,1h),4.72(dd,j=7.9,2.4hz,1h), 3.69(dd,j=11.4,2.8hz,1h),3.54(dd,j=11.3,8.3hz,1h),3.12(br,1h).
13
c nmr(101mhz,cdcl3)δ151.32(dd,j=48.2,12.7hz),148.85(dd,j=47.9,12.7 hz),137.40
–
137.31(m),121.93(dd,j=6.4,3.7hz),117.27(d,j=17.3hz), 115.06(d,j=17.9hz).73.50,67.70.
19
f nmr(376mhz,cdcl3)δ-137.11(ddd,j =20.1,10.9,8.1hz),-138.79(dddd,j=21.5,10.2,7.7,4.3hz).
[0070]
高效液相色谱分离条件:chiracel od-h column,260nm,25℃,n-hexane: i-proh=98:2;流速:1.0ml/min;tr(minor)=20.4min,tr(major)=22.8min.
[0071]
其中,所述中间体进一步应用替格瑞洛重要中间体合成路线如图1;手性化合物iii的hplc谱图如图3;消旋化合物iii的hplc谱图如图4。
[0072]
实施例3-12
[0073]
本发明为考察不对称氢化反应所用催化剂的种类对转化率(conv.)以及对映体选择性(ee)的影响。在实施例2的基础上,将催化剂配体l3依次替换为l1、l2、 l4、l5、l6、l8、l13、l15、l20、l22。
[0074]
实施例3-12中不同催化剂对化合物(ii)还原的转化率以及ee值的影响结果见下表1所示;其中,转化率(conv.)和对映体选择性(ee)由hplc测得。
[0075]
表1
[0076]
编号催化剂反应时间conv.(%)ee(%)实施例2l312h》99》99实施例3l112h9570实施例4l212h9684实施例5l412h9086实施例6l512h》9974实施例7l612h》99》99实施例8l812h》9996实施例9l1312h93》99实施例10l1512h》99》99实施例11l2012h9591实施例12l2212h》9992
[0077]
实施例13-21
[0078]
为考察反应体系所加碱对反应的影响,以异丙醇作为溶剂,在实施例2的基础上,将碳酸钾依次替换为氢氧化钾、碳酸钠、氢氧化钠、叔丁醇钾、叔丁醇钠。反应时间12h,s/c=10000,进行以下实施例13-21,其合成路线如下所示,其结果见下表2所示。
[0079]
表2
[0080][0081][0082]
实施例22-26
[0083]
为考察反应体系溶剂对反应的影响,在实施例2的基础上,将溶剂依次替换为meoh、thf、etoac、dcm、和toluene等。不同溶剂对化合物(ii)还原的转化率以及ee值的影响结果见下表3所示;其中,转化率(conv.)和对映体选择性(ee) 由hplc测得。
[0084]
表3
[0085]
编号催化剂反应溶剂conv.(%)ee(%)
实施例1f-amphox-t
bu-l3iproh》99》99实施例22f-amphox-t
bu-l3meoh4076实施例23f-amphox-t
bu-l3thf8684实施例24f-amphox-t
bu-l3etoac7295实施例25f-amphox-t
bu-l3dcm8266实施例26f-amphox-t
bu-l3toluene9594
[0086]
实施例27-30
[0087]
进一步地,以催化剂f-amphox-t
bu-l3作为催化剂、绿色的溶剂异丙醇作为溶剂,分别改变催化剂用量、反应时间等,结果如下表4所示。
[0088]
表4
[0089]
编号s/c反应温度(℃)反应时间conv.(%)ee(%)实施例2100002512h》99》99实施例2750002512h》99》99实施例28200005012h》99》99实施例29500005024h》99》99实施例301000005024h》99》99
[0090]
实施例31(放大,s/c=100000)
[0091][0092]
在氩气氛围下,将[ir(cod)cl]2(23.9mg,0.0355mmol)和手性配体 f-amphox-t
bu-l3(43.2mg,0.078mmol)溶于20ml异丙醇中,在室温条件下搅拌3小时,得到橙色澄清溶液。用注射器将该橙色溶液加入到中间体(ii)(122 g,710mol)、异丙醇(240ml)和碳酸钾(0.98g,7.1mmol)的混合体系中。将反应体系置于高压釜中,用氢气置换高压釜中的气体三次,最后充入50atm 氢气,在25℃下反应16小时。反应结束后,缓慢释放高压釜中的气体,加入 25ml二氯甲烷,水洗,饱和食盐水洗,无水硫酸钠干燥,减压浓缩得到无色油状液体122g,即氢化产物(iii),产率为99%,经hplc分析,测得ee值为99%。
[0093]
实施例32
[0094][0095]
将(s)-(3,4-二氟苯基)-乙二醇iii(34.8g),二氯甲烷(500ml)投入三口瓶中,搅拌均匀,温度下降到0-5℃,氮气保护下,加入二正丁基锡氧化物(1.2g),对甲苯磺酰氯(45.8g),三乙胺(33.4ml),反应升至室温并在该温度下反应6小时后,用水淬灭反应,分液,水相用二氯甲烷萃取(200ml
×
2),合并有机相,用碳酸氢钠饱和水溶液洗涤,干燥过滤,浓缩后得单磺酰化中间体iv 62.6g,白色固体,收率95%,光学纯98.4%ee。不用纯化,直接用于下步反应。
[0096]
化合物iv数据表征如下:[α]
d25
=+18.6(c=1.00,chcl3).1h nmr(600 mhz,cdcl3)δ7.74(d,j=8.2hz,2h),7.34(d,j=8.1hz,2h),7.15
–
7.08(m,2h), 7.04
–
7.02(m,1h),4.94(dd,j=8.0,3.4hz,1h),4.11(dd,j=10.5,3.5hz,1h), 4.00(dd,j=10.5,8.1hz,1h),2.57(br,1h),2.45(s,3h).
13
c nmr(151mhz, cdcl3)δ151.10(dd,j=29.1,12.7hz),149.45(dd,j=29.1,12.7hz),145.32, 135.41
–
135.35(m),132.37,129.97,127.89,122.26(dd,j=6.5,3.7hz),117.41(d, j=17.4hz),115.32(d,j=18.2hz),73.75,70.79,21.63.
19
f nmr(565mhz, cdcl3)δ-136.68(ddd,j=19.9,10.6,8.3hz),-137.91
–‑
137.98(m).hrms(esi) calculated for c
15h14
f2nao4s[m+na]
+
351.0479;found 351.0470.
[0097]
高效液相色谱分离条件:chiracel od-3column,220nm,30℃,n-hexane: i-proh=90:10;流速:1.0ml/min;tr(minor)=12.0min,tr(major)=14.3min.
[0098]
将上述单磺酰化中间体iv(62.0g)溶于thf中,氮气保护下,体系温度下降到 0-5℃,缓慢加入叔丁醇钾(23.3g),加完后体系温度升至室温,继续反应1小时,用饱和氯化铵溶液淬灭反应,乙酸乙酯稀释后,有机相用水和饱和食盐水洗涤,干燥过滤,浓缩后得到粗品;通过减压蒸馏,收集70-75℃馏分(10mmhg),得到产品即(s)-(3,4-二氟苯基)-环氧乙烷28.3g,收率96%,核磁谱图与文献一致。
[0099]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1