1,5,8-三取代γ-咔啉类衍生物及其制备方法和应用
1,5,8
‑
三取代
γ
‑
咔啉类衍生物及其制备方法和应用
技术领域
1.本发明属药物化学领域,尤其涉及1,5,8
‑
三取代γ
‑
咔啉类衍生物及其制备方法和在制备抗肿瘤药物中的应用。
背景技术:
2.恶性肿瘤是威胁人类健康的重大疾病之一。2020年,全球约1000万人死于癌症,肿瘤的防治一直以来都是各国医药界的重要研究课题。目前临床上常用的抗肿瘤药物普遍存在对实体瘤疗效较差、毒副作用较大、容易产生耐药性等缺点。因此,研究开发更为高效低毒的新型抗肿瘤药物吸引着许多药物研究工作者。咔啉类化合物是一类广泛分布于自然界且具有显著生物活性的三环结构生物碱。根据吡啶环上氮原子的位置不同,可分为α、β、γ、δ
‑
咔啉(o b smirnova,et al,pharm chem j,2011,45:389
‑
400)。一直以来,大量的研究都集中于β
‑
咔啉类化合物(r cao,et al,curr med chem,2007,14:479
‑
500),但γ
‑
咔啉类化合物也具有多样的生物活性,如抗病毒(ksako,et al,bioorg med chem,2008,16:3780
‑
3790)、抗疟原虫(kcimanga,et al,j nat prod,1997,60:688
‑
691)、抗肿瘤(j chen,et al,euro j med chem,2011,46:1343
‑
1347)、抗血栓(cn104211764b)以及中枢神经系统抑制(r e mewshaw,et al,j med chem,1993,36:1488
‑
1495)等活性。此外,该母核也是合成其它药物活性化合物的重要中间体(a molina,et al,j org chem,1996,61:5587
‑
5599)。γ
‑
咔啉独特的结构特征为我们开发一类新型的抗肿瘤化合物提供了新思路。
技术实现要素:
3.本发明的目的在于针对现有技术不足,提供1,5,8
‑
三取代γ
‑
咔啉类衍生物及其制备方法。本发明通过在γ
‑
咔啉的1位引入杂环,并改变8位取代基,合成并筛选出一类新的抗肿瘤化合物。
4.本发明提供的1,5,8
‑
三取代
‑
γ
‑
咔啉类衍生物,其特征在于,它的分子结构如如通式(ⅰ)或通式(ⅱ)所示:
[0005][0006]
其中,
[0007]
a为亚甲基或氧原子;
[0008]
r为氢原子、卤素、甲氧基、甲基或硝基。
[0009]
进一步地,所述的1,5,8
‑
三取代
‑
γ
‑
咔啉类衍生物选自下列任一化合物:5
‑
乙基
‑
1
‑
吗啉
‑8‑
苯甲酰基
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
吗啉
‑8‑
(4
‑
硝基苯甲酰基)
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
哌啶
‑8‑
苯甲酰基
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
硝基苯甲酰基)
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
哌啶
‑8‑
(2
‑
氯苯甲酰基)
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
哌啶
‑8‑
(3
‑
氯苯甲酰基)
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
氯苯甲酰基)
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
氟苯甲酰基)
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
溴苯甲酰基)
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
甲基苯甲酰基)
‑
γ
‑
咔啉、5
‑
乙基
‑1‑
吗啉
‑
n
‑
苯基
‑
γ
‑
咔啉
‑8‑
磺酰胺、5
‑
乙基
‑
n
‑
苯基
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺、n
‑
(4
‑
氯苯基)
‑5‑
乙基
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺、n
‑
(4
‑
溴苯基)
‑5‑
乙基
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺、5
‑
乙基
‑
n
‑
(4
‑
氟苯基)
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺、5
‑
乙基
‑
n
‑
(2
‑
甲氧基苯基)
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺、5
‑
乙基
‑
n
‑
(3
‑
甲氧基苯基)
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺、5
‑
乙基
‑
n
‑
(4
‑
甲氧基苯基)
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺、n
‑
(3,4
‑
二甲氧基苯基)
‑5‑
乙基
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺、5
‑
乙基
‑1‑
哌啶
‑
n
‑
(3,4,5
‑
三甲氧基苯基)γ
‑
咔啉
‑8‑
磺酰胺。
[0010]
本发明的另一个目的是提供该1,5,8
‑
三取代
‑
γ
‑
咔啉类衍生物的制备方法,通过以下步骤实现:
[0011]
所述分子结构通式(ⅰ)的化合物合成路线如下,反应式1:
[0012][0013]
上述反应式用于制备目标产物i。以4
‑
羟基吡啶
‑
2(1h)
‑
酮和苯肼为原料,二苯醚为溶剂,高温反应4h,制备得到化合物2;化合物2经三氯氧磷氯代制得重要中间体3,,进一步在二甲基甲酰胺溶剂中,及在碱性物质催化下与溴乙烷在0
‑
25℃条件下反应,制得化合物4。化合物4与酰氯在氮气保护下进行付克酰基化反应后,经简单后处理即可直接与杂环胺在180℃条件下闷罐反应,制得目标产物i。
[0014]
所述分子结构通式(ⅱ)的化合物合成路线如下,反应式2:
[0015][0016]
上述反应式用于制备目标产物ii。以自制的1
‑
氯
‑5‑
乙基
‑
γ
‑
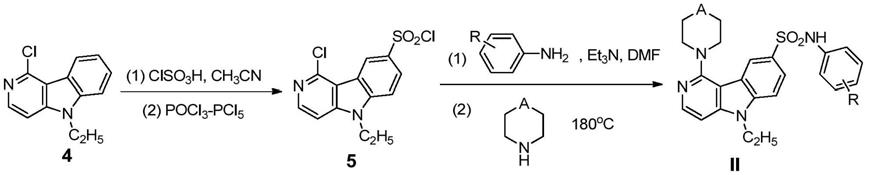
咔啉(4)为原料,与氯磺酸在溶剂乙腈中室温反应2h,制备得到的化合物经三氯氧磷和五氯化磷氯代,制得化合物5;化合物5与芳香胺反应,反应溶剂为二甲基甲酰胺,以三乙胺催化反应,反应时间一般在0.5
‑
2小时,得到的产物经简单后处理即可直接与杂环胺在180℃条件下闷罐反应,制
得目标产物ii。
[0017]
本发明的又一个目的是提供1,5,8
‑
三取代γ
‑
咔啉类衍生物在制备抗肿瘤药物中的应用。初步的体外筛选发现它们对多种肿瘤细胞株,包括人乳腺癌细胞mcf7、人胰腺癌细胞panc
‑
1、人乳腺癌细胞mda
‑
mb
‑
231、人皮肤鳞癌细胞a431、人非小细胞肺癌nci
‑
h1975、人肺癌细胞a549和人神经胶质瘤u87mg有明显的抑制作用,部分化合物对各种肿瘤细胞株的半数抑制浓度(ic
50
)均达到μm级。
[0018]
本发明的特点是重要中间体1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉的合成方法新颖,原料易得,操作简便,为γ
‑
咔啉类化合物的衍生化改造提供了物质基础。本发明的又一个特点是以具有抗肿瘤活性的γ
‑
咔啉为先导化合物,在其1位引入杂环,得到一类结构全新的化合物。初步的药理活性试验表明多数化合物对肿瘤细胞有体外抑制作用,有望用于制备相关的癌症治疗药物,同时也为同类化合物的合成和开发提供新思路。
具体实施方式
[0019]
下面结合具体实施方式对本发明所述的技术方案做进一步说明。
[0020]
本发明公开了1,5,8
‑
三取代
‑
γ
‑
咔啉类衍生物的制备方法,通过以下步骤实现:
[0021]
所述分子结构通式(ⅰ)的化合物合成路线如下:
[0022][0023]
具体包括以下子步骤:
[0024]
(1.1)以4
‑
羟基吡啶
‑
2(1h)
‑
酮和苯肼为原料,二苯醚为溶剂,所述4
‑
羟基吡啶
‑
2(1h)
‑
酮、苯肼、二苯醚三者的物质的量比为1:2~4:9~15,180~240℃反应3~6h,制备得到化合物2;
[0025]
(1.2)步骤(1.1)制得的化合物2经pocl3氯代,所述化合物2与pocl3的物质的量比为1:15~25,回流反应11~15h,制备得到化合物3;
[0026]
(1.3)将体积浓度为50%的dmf
‑
thf混合液与步骤(1.2)制得的化合物3混合,所述化合物3与dmf
‑
thf混合液的比例为1g:8ml~15ml,加入nah和c2h5br,所述nah、c2h5br和化合物3的物质的量比为2~1:1.5~1:1,制备得到化合物4;
[0027]
(1.4)将步骤(1.3)制备得到的化合物4溶于ch2cl2中,化合物与ch2cl2的比例为1mmol/20ml~1mmol/10ml,加入取代的苯甲酰氯和alcl3进行傅克酰基化反应,所述化合物4、取代的苯甲酰氯和alcl3的物质的量比为1:1.5~3:4~6;再与杂环胺在140~180℃条件下闷罐反应,制得目标产物i;
[0028]
所述分子结构通式(ⅱ)的化合物合成路线如下:
[0029][0030]
具体包括以下子步骤:
[0031]
(2.1)以步骤(1.3)制得的化合物4为原料,经磺酸化、氯代制得化合物5;
[0032]
(2.2)将步骤(2.1)制得的化合物5与芳香胺进行缩合反应,再与杂环胺在140~180℃条件下闷罐反应,制得目标产物ii;
[0033]
上述反应式用于制备目标产物ii。以1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉(4)为原料,与氯磺酸在溶剂乙腈中室温反应2h,制备得到的化合物经三氯氧磷和五氯化磷氯代,制得化合物5;化合物5与芳香胺反应,反应溶剂为二甲基甲酰胺,以三乙胺催化反应,反应时间一般在0.5
‑
2小时,得到的产物经简单后处理即可直接与杂环胺在140~180℃条件下闷罐反应,制得目标产物ii。
[0034]
实施例1:制备目标产物i
[0035]
(1.1)以4
‑
羟基吡啶
‑
2(1h)
‑
酮和苯肼为原料,二苯醚为溶剂,所述4
‑
羟基吡啶
‑
2(1h)
‑
酮、苯肼、二苯醚三者的物质的量比为1:2:9,180℃反应3h,制备得到化合物2;
[0036]
(1.2)步骤(1.1)制得的化合物2经pocl3氯代,所述化合物2与pocl3的物质的量比为1:15,回流反应11h,制备得到化合物3;
[0037]
(1.3)将体积浓度为50%的dmf
‑
thf混合液与步骤(1.2)制得的化合物3混合,所述化合物3与dmf
‑
thf混合液的比例为1g:8ml,加入nah和c2h5br,所述nah、c2h5br和化合物3的物质的量比为2:1.5:1,制备得到化合物4;
[0038]
(1.4)将步骤(1.3)制备得到的化合物4溶于ch2cl2中,化合物与ch2cl2的比例为1mmol/10ml,加入取代的苯甲酰氯和alcl3进行傅克酰基化反应,所述化合物4、取代的苯甲酰氯和alcl3的物质的量比为1:1.5:4;再与杂环胺在140℃条件下闷罐反应,制得目标产物i;
[0039]
实施例2:制备目标产物i
[0040]
(1.1)以4
‑
羟基吡啶
‑
2(1h)
‑
酮和苯肼为原料,二苯醚为溶剂,所述4
‑
羟基吡啶
‑
2(1h)
‑
酮、苯肼、二苯醚三者的物质的量比为1:4:15,240℃反应6h,制备得到化合物2;
[0041]
(1.2)步骤(1.1)制得的化合物2经pocl3氯代,所述化合物2与pocl3的物质的量比为1:25,回流反应15h,制备得到化合物3;
[0042]
(1.3)将体积浓度为50%的dmf
‑
thf混合液与步骤(1.2)制得的化合物3混合,所述化合物3与dmf
‑
thf混合液的比例为1g:15ml,加入nah和c2h5br,所述nah、c2h5br和化合物3的物质的量比为1:1:1,制备得到化合物4;
[0043]
(1.4)将步骤(1.3)制备得到的化合物4溶于ch2cl2中,化合物与ch2cl2的比例为1mmol/20ml,加入取代的苯甲酰氯和alcl3进行傅克酰基化反应,所述化合物4、取代的苯甲酰氯和alcl3的物质的量比为1:3:6;再与杂环胺在180℃条件下闷罐反应,制得目标产物i;
[0044]
实施例3:制备目标产物i
[0045]
(1.1)以4
‑
羟基吡啶
‑
2(1h)
‑
酮和苯肼为原料,二苯醚为溶剂,所述4
‑
羟基吡啶
‑
2(1h)
‑
酮、苯肼、二苯醚三者的物质的量比为1:3:12,200℃反应4h,制备得到化合物2;
[0046]
(1.2)步骤(1.1)制得的化合物2经pocl3氯代,所述化合物2与pocl3的物质的量比为1:20,回流反应13h,制备得到化合物3;
[0047]
(1.3)将体积浓度为50%的dmf
‑
thf混合液与步骤(1.2)制得的化合物3混合,所述化合物3与dmf
‑
thf混合液的比例为1g:12ml,加入nah和c2h5br,所述nah、c2h5br和化合物3的物质的量比为1.5:1.2:1,制备得到化合物4;
[0048]
(1.4)将步骤(1.3)制备得到的化合物4溶于ch2cl2中,化合物与ch2cl2的比例为1mmol/18ml,加入取代的苯甲酰氯和alcl3进行傅克酰基化反应,所述化合物4、取代的苯甲酰氯和alcl3的物质的量比为1:1.8:5;再与杂环胺在160℃条件下闷罐反应,制得目标产物i;
[0049]
实施例4:合成化合物:5
‑
乙基
‑1‑
吗啉
‑8‑
苯甲酰基
‑
γ
‑
咔啉(ia)
[0050]
(1.1)合成化合物:(2h)
‑
γ
‑
咔啉
‑1‑
酮(2)
[0051][0052]
在2l三颈瓶中加入4
‑
羟基吡啶
‑
2(1h)
‑
酮(97.2g),苯肼(256.6ml)和二苯醚(936ml),n2保护下搅拌升温至180℃,分水1h,再升至240℃回流反应3h,有固体析出。反应结束后将反应液冷却至室温,抽滤,滤液用甲苯洗。固体用1倍量甲醇重结晶,抽滤,干燥,得黄褐色固体2,收率61%。1h nmr(500mhz,cdcl3):δ=11.87(s,nh),8.41(d,j=8.5hz,1h),8.08(s,1h),7.74(d,j=8.5hz,1h),7.50(d,j=5.0hz,1h),6.95(dd,j=8.5hz,2.0hz,1h),6.89(dd,j=8.5hz,2.0hz,1h),5.91(d,j=5.0hz,1h)。
[0053]
(1.2)合成化合物:1
‑
氯
‑
γ
‑
咔啉(3)
[0054][0055]
在2l的三颈瓶中加入pocl3(1.5l)和化合物2(150g),搅拌溶解后,逐渐升温至115℃,回流反应13h。蒸去pocl3,向残渣中加冰水,控温20℃以下加氨水调至碱性。抽滤,干燥,得淡黄色固体3,收率37.8%。1h nmr(500mhz cdcl3):δ=10.45(s,nh),8.46(d,j=5.0hz,1h),8.12(d,j=8.5hz,1h),8.00(d,j=5.0hz,1h),7.63(d,j=8.5hz,1h),7.50(dd,j=8.5hz,2.0hz,1h),7.29(dd,j=8.5hz,2.0hz,1h)。
[0056]
(1.3)合成化合物:1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉(4)
[0057][0058]
在三颈瓶中加入dmf
‑
thf混合液940ml(v/v=1:1),降温至0℃,加入化合物3
(100g),搅拌使溶解,分批加入nah(29.56g),保持10℃以下搅拌30min,滴加c2h5br(64.43g),30℃反应1.5h。加冰水淬灭,回收thf,残余物用etoac萃取3次,饱和食盐水洗,无水na2so4干燥,回收。柱层析(石油醚:乙酸乙酯:乙醇=10:10:1)得白色固体4,收率70%。1h nmr(500mhz,cdcl3):8.42(d,j=5.0hz,1h),8.14(d,j=2.0hz,1h),7.87(d,j=5.0hz,1h),7.55(d,j=8.5hz,1h),7.40(dd,j=8.5,2.0hz,2h),4.31(q,j=7.0hz,ch2),1.43(t,j=7.0hz,ch3)。
[0059]
(1.4)合成化合物:5
‑
乙基
‑1‑
吗啉
‑8‑
苯甲酰基
‑
γ
‑
咔啉(ia)
[0060][0061]
将1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉(4,0.6mmol)溶于ch2cl2(10ml)中,磁力搅拌下加入苯甲酰氯(0.12mmol)和alcl3(2.7mmol),n2保护下加热回流17h。反应结束后,往反应瓶内加10%naoh,用ch2cl2萃取3次,饱和食盐水洗涤,无水na2so4干燥。过滤,滤液减压回收,粗品用乙酸乙酯润洗,抽滤,干燥,得到的产物与吗啉(10ml)同置于闷罐中,马夫炉180℃恒温反应8h,反应结束后回收反应液,得浅黄色固体。将粗品用ch2cl2润洗,抽滤,干燥,得浅黄色固体,即目标产物ia,收率86%。1h nmr(500mhz,cdcl3):8.41(s,1h),8.32(d,j=5.6hz,1h),8.15(d.j=8.0hz,1h),7.85(d,j=8.0hz,2h),7.54(d,j=8.0hz,1h),7.55(t.j=8.0hz,3h),7.09(d,j=5.6hz,1h),4.44(q,j=7.2hz,ch2),3.76(t,j=4.0hz,4h),3.41(t,j=4.0hz,4h),1.51(t,j=7.2hz,ch3)。
[0062]
实施例5:合成化合物:5
‑
乙基
‑1‑
吗啉
‑8‑
(4
‑
硝基苯甲酰基)
‑
γ
‑
咔啉(ib)
[0063]
步骤(1.1)~步骤(1.3)与实施例4中的步骤(1.1)~步骤(1.3)相同。
[0064]
(1.4)合成化合物:5
‑
乙基
‑1‑
吗啉
‑8‑
(4
‑
硝基苯甲酰基)
‑
γ
‑
咔啉(ib)
[0065][0066]
步骤(1.4)的操作方法与实施例4相同,只是用对硝基苯甲酰氯代替苯甲酰氯,具体为:
[0067]
将1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉(4,0.6mmol)溶于ch2cl2(10ml)中,磁力搅拌下加入对硝基苯甲酰氯(0.12mmol)和alcl3(2.7mmol),n2保护下加热回流17h。反应结束后,往反应瓶内加10%naoh,用ch2cl2萃取3次,饱和食盐水洗涤,无水na2so4干燥。过滤,滤液减压回收,粗品用乙酸乙酯润洗,抽滤,干燥,得到的产物与吗啉(10ml)同置于闷罐中,马夫炉180℃恒温反应8h,反应结束后回收反应液,得浅黄色固体。将粗品用ch2cl2润洗,抽滤,干燥,得浅黄色固体,即目标产物ib,收率47%。1h nmr(500mhz,cdcl3):8.46(s,1h),8.40(d,j=8.4hz,
2h),8.34(d,j=6.0hz,1h),8.03(d,j=8.4hz,1h),7.99(d,j=8.4hz,2h),7.56(d,j=8.4hz,1h),7.11(d,j=6.0hz,1h),4.45(q,j=7.2hz,ch2),3.83(t,j=4.0hz,4h),3.43(t,j=4.0hz,4h),1.52(t,j=7.2hz,ch3).
[0068]
实施例6:合成化合物5
‑
乙基
‑1‑
哌啶
‑8‑
苯甲酰基
‑
γ
‑
咔啉(ic)
[0069]
步骤(1.1)~步骤(1.3)与实施例4中的步骤(1.1)~步骤(1.3)相同。
[0070]
(1.4)合成化合物:5
‑
乙基
‑1‑
哌啶
‑8‑
苯甲酰基
‑
γ
‑
咔啉(ic)
[0071][0072]
步骤(1.4)的操作方法同实施例4,只是用哌啶代替吗啉,具体为:
[0073]
将1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉(4,0.6mmol)溶于ch2cl2(10ml)中,磁力搅拌下加入苯甲酰氯(0.12mmol)和alcl3(2.7mmol),n2保护下加热回流17h。反应结束后,往反应瓶内加10%naoh,用ch2cl2萃取3次,饱和食盐水洗涤,无水na2so4干燥。过滤,滤液减压回收,粗品用乙酸乙酯润洗,抽滤,干燥,得到的产物与哌啶(10ml)同置于闷罐中,马夫炉180℃恒温反应8h,反应结束后回收反应液,得浅黄色固体。将粗品用ch2cl2润洗,抽滤,干燥,得浅黄色固体,即目标产物ic,收率47%。1h nmr(500mhz,cdcl3):8.50(s,1h),8.30(d,j=5.6hz,1h),8.12(d,j=8.0hz,1h),7.86(d,j=7.2hz,2h),7.54(m,4h),7.03(d,j=5.6hz,1h),4.41(q,j=7.2hz,ch2),3.33(m,4h),2.05(m,4h),1.90(m,2h),1.50(t,j=7.2hz,ch3).
[0074]
实施例7:合成化合物:5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
硝基苯甲酰基)
‑
γ
‑
咔啉(id)
[0075]
步骤(1.1)~步骤(1.3)与实施例4中的步骤(1.1)~步骤(1.3)相同。
[0076]
(1.4)合成化合物5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
硝基苯甲酰基)
‑
γ
‑
咔啉(id)
[0077][0078]
步骤(1.4)的操作方法同实施例4,只是用对硝基苯甲酰氯代替苯甲酰氯,用哌啶代替吗啉,具体为:
[0079]
将1
‑
氯
‑5‑
乙基
‑
基
‑
咔啉(4,0.6mmol)溶于ch2cl2(10ml)中,磁力搅拌下加入对硝基苯甲酰氯(0.12mmol)和alcl3(2.7mmol),n2保护下加热回流17h。反应结束后,往反应瓶内加10%naoh,用ch2cl2萃取3次,饱和食盐水洗涤,无水na2so4干燥。过滤,滤液减压回收,粗品用乙酸乙酯润洗,抽滤,干燥,得到的产物与哌啶(10ml)同置于闷罐中,马夫炉180℃恒温反应8h,反应结束后回收反应液,得浅黄色固体。将粗品用ch2cl2润洗,抽滤,干燥,得浅黄色固体,即目标产物id,收率63%。1h nmr(500mhz,cdcl3):8.47(d,j=1.2hz,1h),8.39(d,j=8.4hz,2h),8.32(d,j=6.0hz,1h),8.08(dd,j=1.2,8.4hz,1h),7.99(d,j=8.4hz,2h),7.54(d,j=8.4hz,1h),7.04(d,j=6.0hz,1h),4.43(q,j=7.2hz,ch2),3.38(m,4h),1.61
(m,4h),1.50(t,j=7.2hz,ch3),1.26(m,2h)。
[0080]
实施例8:合成化合物:5
‑
乙基
‑1‑
哌啶
‑8‑
(2
‑
氯苯甲酰基)
‑
γ
‑
咔啉(ie)
[0081]
步骤(1.1)~步骤(1.3)与实施例4中的步骤(1.1)~步骤(1.3)相同。
[0082]
(1.4)合成化合物:5
‑
乙基
‑1‑
哌啶
‑8‑
(2
‑
氯苯甲酰基)
‑
γ
‑
咔啉(ie)
[0083][0084]
步骤(1.4)的操作方法同实施例4,只是用邻氯苯甲酰氯代替苯甲酰氯,用哌啶代替吗啉,具体为:
[0085]
将1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉(4,0.6mmol)溶于ch2cl2(10ml)中,磁力搅拌下加入邻氯苯甲酰氯(0.12mmol)和alcl3(2.7mmol),n2保护下加热回流17h。反应结束后,往反应瓶内加10%naoh,用ch2cl2萃取3次,饱和食盐水洗涤,无水na2so4干燥。过滤,滤液减压回收,粗品用乙酸乙酯润洗,抽滤,干燥,得到的产物与哌啶(10ml)同置于闷罐中,马夫炉180℃恒温反应8h,反应结束后回收反应液,得浅黄色固体。将粗品用ch2cl2润洗,抽滤,干燥,得浅黄色固体,即目标产物ie,收率88%。1h nmr(500mhz,cdcl3):8.43(s,1h),8.29(d,j=6.0hz,1h),8.17(dd,j=9.0,1.2hz,1h),7.49(m,3h),7.13(t,j=7.2hz,2h),7.02(d,j=6.0hz,1h),4.40(q,j=7.2hz,ch2),3.28(m,4h),2.93(t,j=4.8hz,4h),1.55(m,2h),1.49(t,j=7.2hz,ch3)。
[0086]
实施例9:合成化合物:5
‑
乙基
‑1‑
哌啶
‑8‑
(3
‑
氯苯甲酰基)
‑
γ
‑
咔啉(if)
[0087]
步骤(1.1)~步骤(1.3)与实施例4中的步骤(1.1)~步骤(1.3)相同。
[0088]
(1.4)合成化合物:5
‑
乙基
‑1‑
哌啶
‑8‑
(3
‑
氯苯甲酰基)
‑
γ
‑
咔啉(if)
[0089][0090]
操作方法同实施例4,只是用间氯苯甲酰氯代替苯甲酰氯,用哌啶代替吗啉,具体为:
[0091]
将1
‑
氯
‑5‑
乙基
‑
基
‑
咔啉(4,0.6mmol)溶于ch2cl2(10ml)中,磁力搅拌下加入间氯苯甲酰氯(0.12mmol)和alcl3(2.7mmol),n2保护下加热回流17h。反应结束后,往反应瓶内加10%naoh,用ch2cl2萃取3次,饱和食盐水洗涤,无水na2so4干燥。过滤,滤液减压回收,粗品用乙酸乙酯润洗,抽滤,干燥,得到的产物与哌啶(10ml)同置于闷罐中,马夫炉180℃恒温反应8h,反应结束后回收反应液,得浅黄色固体。将粗品用ch2cl2润洗,抽滤,干燥,得浅黄色固体,即目标产物if,收率82%。1h nmr(500mhz,cdcl3):8.49(d,j=1.8hz,1h),8.33(d,j=
6.0hz,1h),8.13(dd,j=8.4,1.8hz,1h),7.84(t,j=1.8hz,1h),7.76(td,j=7.8,1.8hz,1h),7.61(td,j=7.8,1.8hz,1h),7.55(d,j=8.4hz,1h),7.50(t,j=7.8hz,1h),7.05(d,j=6.0hz,1h),4.44(q,j=7.2hz,ch2),3.37(m,4h),1.68(m,4h),1.63(m,2h),1.51(t,j=7.2hz,ch3).
[0092]
实施例10:合成化合物5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
氯苯甲酰基)
‑
γ
‑
咔啉(ig)
[0093][0094]
操作方法同实施例4,只是在步骤(1.4)中用对氯苯甲酰氯代替苯甲酰氯,用哌啶代替吗啉,得浅黄色固体,即目标产物ig,收率86%。1h nmr(500mhz,cdcl3):8.53(d,j=1.2hz,1h),8.30(d,j=6.0hz,1h),8.03(dd,j=1.2,8.4hz,1h),7.87(dd,j=1.8,7.2hz,2h),7.51(d,j=8.4hz,1h),7.03(d,j=6.0hz,1h),6.95(d,j=1.8,7.2hz,2h),4.41(q,j=7.2hz,ch2),3.40(m,4h),1.77(m,4h),1.64(m,2h),1.48(t,j=7.2hz,ch3).
[0095]
实施例11:合成化合物5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
氟苯甲酰基)
‑
γ
‑
咔啉(ih)
[0096][0097]
操作方法同实施例4,只是在步骤(1.4)中用对氟苯甲酰氯代替苯甲酰氯,用哌啶代替吗啉,得浅黄色固体,即目标产物ih,收率88%。1h nmr(500mhz,cdcl3):8.53(d,j=1.2hz,1h),8.31(d,j=6.0hz,1h),8.04(dd,j=1.2,8.4hz,1h),7.88(d,j=8.4hz,2h),7.51(d,j=8.4hz,1h),7.04(d,j=6.0hz,1h),6.96(d,j=8.4hz,2h),4.42(q,j=7.2hz,ch2),3.41(m,4h),1.77(m,4h,),1.64(m,2h),1.50(t,3h,j=7.2hz,ch3).
[0098]
实施例12:合成化合物5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
溴苯甲酰基)
‑
γ
‑
咔啉(ii)
[0099][0100]
操作方法同实施例4,只是在步骤(1.4)中用对溴苯甲酰氯代替苯甲酰氯,用哌啶代替吗啉,得浅黄色固体,即目标产物ii,收率63%。1h nmr(500mhz,cdcl3):8.53(s,1h),8.31(d,j=6.0hz,1h),8.04(d,j=8.4hz,1h),7.88(d,j=8.4hz,2h),7.51(d,j=8.4hz,1h),7.04(d,j=6.0hz,1h),6.96(d,j=8.4hz,2h),4.42(q,j=7.2hz,ch2),3.41(m,4h),1.80(m,4h),1.64(m,2h),1.50(t,j=7.2hz,ch3).
[0101]
实施例13:合成化合物5
‑
乙基
‑1‑
哌啶
‑8‑
(4
‑
甲基苯甲酰基)
‑
γ
‑
咔啉(ij)
[0102][0103]
操作方法同实施例4,只是在步骤(1.4)中用对甲基苯甲酰氯代替苯甲酰氯,用哌啶代替吗啉,得浅黄色固体,即目标产物ij,收率42%。1h nmr(500mhz,cdcl3):8.51(s,1h),8.30(d,j=6.0hz,1h),8.08(dd,j=1.2,8.4hz,1h),7.78(d,j=8.4hz,2h),7.50(d,j=8.4hz,1h),7.32(d,j=8.4hz,2h),7.02(d,j=6.0hz,1h),4.41(q,j=7.2hz,ch2),3.41(m,4h),2.47(s,ch3),1.67(m,4h),1.60(m,2h),1.48(t,j=7.2hz,ch3).
[0104]
实施例14:合成化合物:5
‑
乙基
‑1‑
吗啉
‑
n
‑
苯基
‑
γ
‑
咔啉
‑8‑
磺酰胺(iia)
[0105]
(2.1)合成化合物:1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉
‑8‑
磺酰氯(5)
[0106][0107]
将化合物4(30g)溶于乙腈(300ml),降温至0℃以下,滴加hso3cl(91.2g),室温反应2d。反应结束,将反应液倒入3倍量冰水中,析出黄色固体,抽滤,固体用丙酮润洗,干燥得类白色固体1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉
‑8‑
磺酸。将所得的化合物(35g),pcl5(93.86g)和pocl3(4.34g)混合均匀,90℃反应4h。反应完毕后将反应物倒入冰水中,缓慢加入饱和碳酸钠溶液中和,用ch2cl2萃取,饱和食盐水洗2次,无水硫酸钠干燥,回收得白色固体5,收率70%。1h nmr(500mhz,cdcl3):8.22(d,j=5.0hz,1h),7.95(d,j=8.5hz,1h),7.84(d,j=2.0hz,1h),7.80(dd,j=8.5,2.0hz,1h),7.67(d,j=5.0hz,1h),4.31(q,j=7.0hz,ch2),1.43(t,j=7.0hz,ch3)。
[0108]
(2.2)合成化合物5
‑
乙基
‑1‑
吗啉
‑
n
‑
苯基
‑
γ
‑
咔啉
‑8‑
磺酰胺(iia)
[0109][0110]
将1
‑
氯
‑5‑
乙基
‑
γ
‑
咔啉
‑8‑
磺酰氯(0.2mmol)溶于dmf(1ml)中,室温搅拌下加入苯胺(0.2mmol)和et3n(0.4mmol),继续室温搅拌1h。反应结束后,往反应瓶内加水淬灭,使反应终止。反应液抽滤,粗品用ch2cl2润洗,抽滤,干燥,得到的白色固体与吗啉(4ml)同置于闷罐中,马夫炉180℃恒温反应8h,反应结束后回收反应液,得浅黄色固体。将粗品用ch2cl2润洗,抽滤,干燥,得白色固体,即目标产物iia,收率76%。1h nmr(500mhz,dmso
‑
d6):10.22(br s,1h),8.25(d,j=6.0hz,1h),8.22(s,1h),7.92(m,2h),7.40(d,j=6.0hz,1h),7.21
(t,j=8.0hz,2h),7.11(d,j=8.0hz,2h),6.96(t,j=8.0hz,1h),4.49(q,j=7.2hz,ch2),3.87(t,j=4.4hz,4h),3.25(t,j=4.4hz,4h),1.32(t,j=7.2hz,ch3).
[0111]
实施例15:合成化合物5
‑
乙基
‑
n
‑
苯基
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺(iib)
[0112][0113]
操作方法同实施例14,只是在步骤(2.1)中用哌啶代替吗啉,得白色固体,即目标产物iib,收率99%。1h nmr(500mhz,dmso
‑
d6):10.13(s,1h),8.29(s,1h),8.22(d,j=5.6hz,1h),7.90(m,2h),7.33(d,j=5.6hz,1h),7.19(t,j=8.0hz,2h),7.12(d,j=8.0hz,2h),6.97(t,j=8.0hz,1h),4.48(q,j=7.2hz,ch2),3.23(br s,4h),1.70(m,4h),1.69(m,2h),1.31(t,j=7.2hz,ch3).
[0114]
实施例16:合成化合物n
‑
(4
‑
氯苯基)
‑5‑
乙基
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺(iic)
[0115][0116]
操作方法同实施例14,只是在步骤(2.1)中用对氯苯胺代替苯胺,用哌啶代替吗啉,得白色固体,即目标产物iic,收率87%。1h nmr(500mhz,dmso
‑
d6):9.25(brs,1h),8.27(d,j=1.8hz,1h),8.22(d,j=5.4hz,1h),7.90(dd,j=1.8,8.4hz,1h),7.87(d,j=8.4hz,1h),7.33(d,j=5.4hz,1h),7.27(d,j=9.6hz,2h),7.15(d,j=9.6hz,2h),4.51(q,j=7.2hz,ch2),3.23(br s,4h),1.82(m,4h),1.68(m,2h),1.36(t,j=7.2hz,ch3).
[0117]
实施例17:合成化合物n
‑
(4
‑
溴苯基)
‑5‑
乙基
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺(iid)
[0118][0119]
操作方法同实施例14,只是在步骤(2.1)中用对溴苯胺代替苯胺,用哌啶代替吗啉,得白色固体,即目标产物iid,收率84%。1h nmr(500mhz,dmso
‑
d6):8.75(brs,1h),8.26(s,1h),8.23(d,j=5.6hz,1h),7.87(m,2h),7.39(d,j=8.8hz,2h),7.34(d,j=5.6hz,1h),7.08(d,j=8.8hz,2h),4.49(q,j=7.2hz,ch2),3.24(br s,4h),1.69(m,4h),1.66(m,2h),1.32(t,j=7.2hz,ch3).
[0120]
实施例18:合成化合物5
‑
乙基
‑
n
‑
(4
‑
氟苯基)
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺(iie)
[0121][0122]
操作方法同实施例14,只是在步骤(2.1)中用对氟苯胺代替苯胺,用哌啶代替吗啉,得白色固体,即目标产物iie,收率25%。1h nmr(500mhz,dmso
‑
d6):8.48(brs,1h),8.35(d,j=1.2hz,1h),8.24(d,j=5.6hz,1h),8.09(m,1h),7.90(dd,j=1.2,8.8hz,1h),7.86(d,j=8.8hz,1h),7.36(d,j=5.6hz,1h),7.25(m,1h),7.03(t,j=8.8hz,2h),4.52(q,j=7.2hz,ch2),3.32(br s,4h),1.82(m,4h),1.68(m,2h),1.35(t,j=7.2hz,ch3).
[0123]
实施例19:合成化合物5
‑
乙基
‑
n
‑
(2
‑
甲氧基苯基)
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺(iif)
[0124][0125]
操作方法同实施例14,只是在步骤(2.1)中用2
‑
甲氧基苯胺代替苯胺,用哌啶代替吗啉,得白色固体,即目标产物iif,收率43%。1h nmr(500mhz,dmso
‑
d6):9.21(brs,1h),8.19(d,j=6.0hz,1h),8.16(s,1h),7.87(dd,j=2.0,8.5hz,1h),7.83(d,j=8.5hz,1h),7.32(d,j=6.0hz,1h),7.28(dd,j=8.0,1.0hz,1h),7.03(t,j=8.0hz,1h),6.84(t,j=8.0hz,1h),6.79(d,j=8.0hz,1h),4.47(q,j=7.0hz,ch2),3.30(s,3h),3.17(br s,4h),1.69(m,4h),1.62(m,2h),1.29(t,j=7.0hz,ch3).
[0126]
实施例20:合成化合物5
‑
乙基
‑
n
‑
(3
‑
甲氧基苯基)
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺(iig)
[0127][0128]
操作方法同实施例14,只是在步骤(2.1)中用3
‑
甲氧基苯胺代替苯胺,用哌啶代替吗啉,得白色固体,即目标产物iig,收率80%。1h nmr(500mhz,dmso
‑
d6):10.26(brs,1h),8.33(d,j=1.2hz,1h),8.22(d,j=6.0hz,1h),7.91(dd,j=1.2,8.4hz,1h),7.87(d,j=8.4hz,1h),7.33(d,j=6.0hz,1h),7.08(t,j=8.4hz,1h),6.73(d,j=2.4hz,1h),6.71(d,j=8.4hz,1h),6.53(dd,j=2.4,8.4hz,1h),4.47(q,j=7.2hz,ch2),3.62(s,och3),3.25(br s,4h),1.81(m,4h),1.68(m,2h),1.31(t,j=7.2hz,ch3).
[0129]
实施例21:合成化合物5
‑
乙基
‑
n
‑
(4
‑
甲氧基苯基)
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺
(iih)
[0130][0131]
操作方法同实施例14,只是在步骤(2.1)中用4
‑
甲氧基苯胺代替苯胺,用哌啶代替吗啉,得白色固体,即目标产物iih,收率74%。1h nmr(500mhz,dmso
‑
d6):9.23(brs,1h),8.21(d,j=5.6hz,1h),8.17(s,1h),7.84(m,2h),7.33(d,j=5.6hz,1h),6.99(d,j=8.8hz,2h),6.76(d,j=8.8hz,2h),4.48(q,j=7.2hz,ch2),3.61(s,och3),3.21(br s,4h),1.73(m,4h),1.66(m,2h),1.32(t,j=7.2hz,ch3).
[0132]
实施例22:合成化合物n
‑
(3,4
‑
二甲氧基苯基)
‑5‑
乙基
‑1‑
哌啶
‑
γ
‑
咔啉
‑8‑
磺酰胺(iii)
[0133][0134]
操作方法同实施例14,只是在步骤(2.1)中用3,4
‑
甲氧基苯胺代替苯胺,用哌啶代替吗啉,得白色固体,即目标产物iii,收率31%。1h nmr(500mhz,dmso
‑
d6):9.81(s,1h),8.22(m,2h),7.86(s,2h),7.33(d,j=6.0hz,1h),6.74(m,2h),6.54(d,j=8.4hz,1h),4.49(q,j=7.2hz,ch2),3.60(s,3h,och3),3.58(s,3h,och3),3.20(br s,4h),1.73(m,4h),1.66(m,2h),1.32(t,j=7.2hz,ch3).
[0135]
实施例23:合成化合物5
‑
乙基
‑1‑
哌啶
‑
n
‑
(3,4,5
‑
三甲氧基苯基)γ
‑
咔啉
‑8‑
磺酰胺(iij)
[0136][0137]
操作方法同实施例14,只是在步骤(2.1)中用3,4,5
‑
甲氧基苯胺代替苯胺,用哌啶代替吗啉,得白色固体,即目标产物iij,收率67%。1h nmr(500mhz,dmso
‑
d6):8.60(brs,1h),8.29(d,j=1.2hz,1h),8.23(d,j=5.4hz,1h),7.92(dd,j=1.2,9.0hz,1h),7.89(d,j=9.0hz,1h),7.35(d,j=5.4hz,1h),6.39(s,2h),4.49(q,j=7.2hz,ch2),3.50(s,3h,och3),3.32(s,3h,och3),3.23(s,3h,och3),3.01(t,j=5.4hz,4h),1.77(m,4h),1.56(m,2h),1.32(t,j=7.2hz,ch3).
[0138]
实施例24:γ
‑
咔啉类衍生物对不同肿瘤细胞的体外抑制作用
[0139]
以人乳腺癌细胞mcf7、人胰腺癌细胞panc
‑
1、人乳腺癌细胞mda
‑
mb
‑
231、人皮肤鳞癌细胞a431、人非小细胞肺癌nci
‑
h1975、人肺癌细胞a549、人神经胶质瘤u87mg为对象,采用mtt法测试了目标化合物对肿瘤细胞株的抑制作用。将处于对数生长期的肿瘤细胞,以2
×
104个/ml接种于96孔培养板中,每孔加细胞悬液200μl,在培养24h后,分别加入5种浓度的待测化合物(0.08
‑
50μg/ml)2μl,每个浓度设3个复孔。细胞在37℃,5%co2培养箱中孵育72小时后,加入浓度为5mg/ml的mtt溶液10μl,继续培养4小时。吸去上清液,加入100μl dmso摇匀,用酶标仪于570nm波长下测定各孔的od值,细胞抑制率的计算公式为:细胞抑制率%=(对照组od值-用药组od值)/对照细胞od值
×
100%,再用bliss法求出ic50。具体结果(为三次测试的平均值)参见表1。由表1可知,本发明实施例制得的目标产物对多种肿瘤细胞株,包括人乳腺癌细胞mcf7、人胰腺癌细胞panc
‑
1、人乳腺癌细胞mda
‑
mb
‑
231、人皮肤鳞癌细胞a431、人非小细胞肺癌nci
‑
h1975、人肺癌细胞a549、人神经胶质瘤u87mg有明显的抑制作用,部分化合物对各种肿瘤细胞株的半数抑制浓度(ic
50
)均达到μm级。
[0140]
表1:本发明实施例所合成的部分1,5,8
‑
三取代γ
‑
咔啉类衍生物作用72小时对不同肿瘤细胞的体外抑制作用
[0141][0142][0143]
a
“‑”
表示未测定ic
50
值。
[0144]
综上所述,本发明公开的1,5,8
‑
三取代
‑
γ
‑
咔啉类衍生物的合成方法新颖,原料易得,操作简便,为γ
‑
咔啉类化合物的衍生化改造提供了物质基础。本发明的又一个特点是以具有抗肿瘤活性的γ
‑
咔啉为先导化合物,在其1位引入杂环,得到一类结构全新的化合物。初步的药理活性试验表明多数化合物对肿瘤细胞有体外抑制作用,有望用于制备相关的癌症治疗药物,同时也为同类化合物的合成和开发提供新思路。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1