一种钴催化合成喹啉及喹唑啉类化合物的方法
1.本发明涉及一种喹唑啉及其衍生物或喹啉及其衍生物的合成方法,具体涉及一种以醋酸钴为催化剂,无须使用配体、反应条件温和、环保性好、操作简便、成本低、反应收率高的喹唑啉及其衍生物或喹啉及其衍生物的合成方法,属于有机合成技术领域。
背景技术:
2.氮杂环化合物具有良好的药物和生物活性,作为大多数药物分子和天然产物的重要组成骨架,开发并设计高效绿色的合成方法学用于制备氮杂环化合物一直是化学研究的热点领域之一。
3.喹啉及其衍生物是一类重要的含氮杂环化合物。许多药物分子中都含有喹啉结构,具有抗菌、抗疟和抗癌等活性。喹啉衍生物也被广泛用于制备除草剂、缓蚀剂、染料和络合剂。传统的合成方法主要是基于苯胺或其衍生物与甘油或羰基化合物的酸催化缩合。这种方法的缺点是需要使用化学计量的酸催化剂,副产物多且对环境污染较大。酸催化剂的使用会限制喹啉衍生物合成中可能对酸敏感的取代基的引入。此外,喹唑啉类化合物在生物分子和有机功能材料中也扮演着重要角色,许多功能分子以及药物中都有喹唑啉的骨架。传统合成喹唑啉衍生物的方法主要有n
‑
烷基化芳基脒类分子间氧化成环或通过卤苯与酰胺的偶联成环等,但大多数合成方法会产生含卤等废物,原子利用率低,反应效率不高。
4.近年来,基于无受体脱氢偶联(acceptorless dehydrogenative coupling)反应用于合成氮杂环化合物受到了科研工作者的广泛关注,该反应具有反应条件温和、对环境污染小,原料廉价易得等优势,特别是副产物仅为对环境无危害的氢气和水。大量基于钌、铱等贵金属配合物被报道可以高效催化无受体脱氢偶联反应,与此同时人们也开发了一系列高效的铁、钴、镍、锰等廉价过渡金属催化剂。这些廉价金属催化剂虽然催化剂效果比相应的贵金属催化剂略低,但由于其价格低廉也被广泛用于催化脱氢、加氢反应以及n
‑
烷基化等反应。因此开发高活性的廉价过渡金属催化剂,并用于杂环化合物的制备具有十分广阔的应用前景。
5.目前,有采用下述两种路线来制备喹啉类化合物或喹唑啉类化合物的,具体如下表1和表2所示:
从这些现有技术可以看出,无受体脱氢偶联催化体系虽然都能较高效的催化合成喹啉、喹唑啉类化合物,但大多数催化体系为贵金属催化剂,也就是说需要先通过有机合成手段制备得到金属配合物,或使用对空气和湿度敏感的有机膦化合物作为配体,这些方面都限制了其工业应用前景。因此,选择廉价易得的催化剂,在温和条件下实现喹啉及喹唑啉类化合物的合成具有重要实际的意义和研究价值。
技术实现要素:
6.针对现今催化合成喹啉及喹唑啉类化合物技术存在的不足,本发明提供了一种钴催化合成喹啉及喹唑啉类化合物的方法,该方法直接以醋酸钴为催化剂,不需要将其合成相应的配合物,也不需要配体,催化剂体系简单,催化活性高,成本低。
7.本发明具体技术方案如下:一种钴催化合成喹啉类化合物的方法,其特征是:由化合物1与化合物2在催化剂和碱的存在下经无受体脱氢偶联反应得到,所述催化剂为醋酸钴;化合物1结构式如下,式1中,r1在苯环中的位置不固定,r1可以为氢、甲基、氟、氯或
溴,优选的,r1为氢或溴,更优选的,r1为氢。
8.化合物2结构式如下,式2中,r2为甲基、乙基、苯基、4
‑
甲基苯基、4
‑
甲氧基苯基、4
‑
氯苯基、4
‑
溴苯基、3
‑
甲基苯基、3
‑
甲氧基苯基、3
‑
氯苯基、2
‑
甲基苯基、2
‑
氯苯基、2
‑
溴苯基或3
‑
吡啶基,同时r3为氢或甲基;或者,r2、r3共同构成环戊基或环己基。
9.优选的,r2为苯基、4
‑
甲氧基苯基、4
‑
甲基苯基或3
‑
甲基苯基,同时r3为氢。更优选的,r2为苯基,同时r3为氢。
10.所得喹啉类化合物结构式如下,式3中,r1、r2、r3定义同上。
11.化合物1与化合物2的反应式如下:进一步的,上述合成方法中,所述醋酸钴为无水醋酸钴或四水合醋酸钴。醋酸钴与碱搭配可以起到很好的催化效果,无须将其制成金属配合物或者额外加入配体,这大大提高了工艺的操作便捷性,简化了工艺流程,降低了成本。优选的,催化剂的摩尔用量为化合物1的摩尔量的6
‑
10%。
12.进一步的,所述碱为ko
t
bu(叔丁醇钾),采用该碱与醋酸钴搭配时具有更好的转化率。碱的摩尔用量为化合物1的摩尔量的70%
‑
100%。
13.进一步的,化合物1与化合物2的摩尔比为1:1
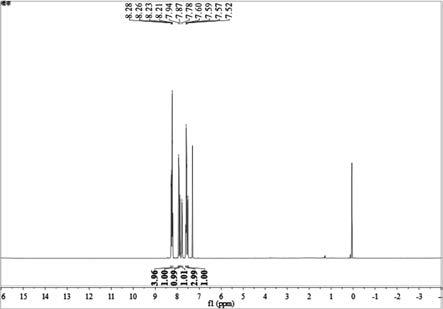
‑
1.2。
14.进一步的,反应在有机溶剂存在下进行,有机溶剂可以为甲苯、四氢呋喃或1, 4
‑
二氧六环,优选的,当有机溶剂为甲苯时效果更佳。
15.进一步的,反应温度为80℃
‑
110℃,在该温度下直至反应完成。反应时间一般为12 h左右。
16.进一步的,反应在气体保护下进行,所述保护性气体为氩气或氮气。
17.本发明还提供了一种钴催化合成喹唑啉类化合物的方法,其由化合物1与化合物4
在催化剂和碱的存在下经无受体脱氢偶联反应得到,所述催化剂为醋酸钴。
18.进一步的,化合物1结构式如下,式1中,r1在苯环中的位置不固定,r1可以为氢、甲基、氟、氯或溴,优选的,r1为氢。
19.进一步的,化合物4结构式如下,式4中,r4为苯基、4
‑
甲基苯基、4
‑
甲氧基苯基、4
‑
氯苯基、4
‑
溴苯基、4
‑
硝基苯基、3
‑
甲基苯基、3
‑
甲氧基苯基、3
‑
氯苯基、2
‑
甲基苯基或2
‑
氯苯基,优选为苯基、4
‑
甲基苯基、4
‑
甲氧基苯基、3
‑
甲氧基苯基、4
‑
溴苯基、3
‑
甲基苯基或2
‑
甲基苯基,更优选为苯基。
20.所得喹唑啉类化合物结构式如下,式5中,r1、r4定义同上。
21.化合物1与化合物4的反应式如下:进一步的,上述喹唑啉类化合物的合成方法中,所述醋酸钴为无水醋酸钴或四水合醋酸钴。醋酸钴与碱搭配可以起到很好的催化效果,无须将其制成金属配合物或者额外加入配体,这大大提高了工艺的操作便捷性,简化了工艺流程,降低了成本。优选的,催化剂的摩尔用量为化合物4的摩尔量的6
‑
10%。
22.进一步的,上述喹唑啉类化合物的合成方法中,所述碱为ko
t
bu(叔丁醇钾),采用该碱与醋酸钴搭配时具有更好的转化率。碱的摩尔用量为化合物4的摩尔量的70%
‑
100%。
23.进一步的,上述喹唑啉类化合物的合成方法中,化合物1与化合物4的摩尔比为1.5
‑
2:1。
24.进一步的,上述喹唑啉类化合物的合成方法中,反应在有机溶剂存在下进行,有机溶剂可以为甲苯、叔戊醇或二甲苯。优选的,当有机溶剂为叔戊醇时效果更佳。
25.进一步的,上述喹唑啉类化合物的合成方法中,反应温度为90
‑
100℃,在该温度下直至反应完成。反应时间一般为24 h左右。
26.进一步的,反应在气体保护下进行,所述保护性气体为氩气或氮气。
27.本发明具有以下有益效果:1、本发明直接以醋酸钴为催化剂高效催化无受体脱氢偶联反应,无须将金属钴制
(134.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水, 并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3b (192.8 mg, 收率88%)。
37.实施例6将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),4
‑
甲氧基苯乙酮2c (150.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3c (211.6 mg, 收率90%)。
38.实施例7将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),4
‑
氯苯乙酮2d (154.6 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3d (198.4 mg, 收率83%)。
39.实施例8将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),4
‑
溴苯乙酮2e (199.0 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3e (240.5 mg, 收率85%)。
40.实施例9将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),3
‑
甲基苯乙酮2f (134.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3f (190.6 mg, 收率87%)。
41.实施例10将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),3
‑
甲氧基苯乙酮2g (150.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml1, 4
‑
二氧六环并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3g (190.4 mg, 收率81%)。
42.实施例11将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),3
‑
氯苯乙酮2h (154.6 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,
并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3h (181.7 mg, 收率76%)。
43.实施例12将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),2
‑
甲基苯乙酮2i (134.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3i (175.3 mg, 收率80%)。
44.实施例13将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),2
‑
氯苯乙酮2j (154.6 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3j (196.0 mg, 收率82%)。
45.实施例14将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),2
‑
溴苯乙酮2k (199.0 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml四氢呋喃并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3k (217.9 mg, 收率77%)。
46.实施例15将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),3
‑
乙酰基吡啶2l (121.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3l (107.2 mg, 收率52%)。
47.实施例16将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),丁酮2m (72.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到黄色油状液体3m (40.8 mg, 收率26%)。
48.实施例17将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),3
‑
戊酮2n (86.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到黄色油状液体3n (87.3 mg, 收率51%)。
49.实施例18将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),环戊酮2o (84.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到黄色油状液体3o (108.2 mg, 收率64%)。
50.实施例19将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),环己酮2p (98.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到黄色油状液体3p (144.6 mg, 收率79%)。
51.实施例20将反应容器中加入2
‑
氨基
‑5‑
氟苯甲醇1q (141.0 mg, 1.0 mmol),苯乙酮2a (120.2 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3q (98.2 mg, 收率44%)。
52.实施例21将反应容器中加入2
‑
氨基
‑5‑
氯苯甲醇1r (157.6 mg, 1.0 mmol),苯乙酮2a (120.2 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固液体3r (179.3 mg, 收率75%)。
53.实施例22将反应容器中加入2
‑
氨基5
‑
溴苯甲醇1s (202.0 mg, 1.0 mmol),苯乙酮2a (120.2 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3s (234.9 mg, 收率83%)。
54.实施例23将反应容器中加入2
‑
氨基
‑5‑
氯苯甲醇1t (157.6 mg, 1.0 mmol),4
‑
甲基苯乙酮2b (134.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3t (194.9 mg, 收率77%)。
55.实施例24
将反应容器中加入2
‑
氨基
‑5‑
氯苯甲醇1u (157.6 mg, 1.0 mmol),4
‑
甲氧基苯乙酮2c (150.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3u (215.2 mg, 收率80%)。
56.实施例25将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),苯甲腈4a (103.1 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5a (195.8 mg, 收率95%),其核磁谱图如图2所示。
57.实施例26将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),苯甲腈4a (103.1 mg, 1.0 mmol),co(oac)2•
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (78.4 mg, 0.7 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5a (154.6 mg, 收率75%)。
58.实施例27将反应容器中加入2
‑
氨基苯甲醇1a (184.7 mg, 1.5 mmol),苯甲腈4a (103.1 mg, 1.0 mmol),co(oac)2·
4h2o(24.9 mg, 0.1 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在90℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5a (187.5 mg, 收率91%)。
59.实施例28将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),4
‑
甲基苯甲腈4b (117.2 mg, 1.0 mmol),co(oac)2·
4h2o(14.9 mg, 0.06 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5b (202.5 mg, 收率92%)。
60.实施例29将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),4
‑
甲氧基苯甲腈4c (133.2 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在100℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5c (212.5 mg, 收率90%)。
61.实施例30将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),4
‑
氯苯甲腈4d (137.6 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5d (180.0 mg, 收率75%)。
62.实施例31将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),4
‑
溴苯甲腈4e (182.0 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的甲苯并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5e (230.0 mg, 收率81%)。
63.实施例32将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),4
‑
硝基苯甲腈4f (148.1 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5f (70.3 mg, 收率28%)。
64.实施例33将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),3
‑
甲基苯甲腈4g (117.2 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5g (195.9 mg, 收率89%)。
65.实施例34将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),3
‑
甲氧基苯甲腈4h (133.2 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的二甲苯并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5h (200.7 mg, 收率85%)。
66.实施例35将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),3
‑
氯苯甲腈4i (137.6 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml
水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5i (177.6 mg, 收率74%)。
67.实施例36将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),2
‑
甲基苯甲腈4j (117.2 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5j (187.1 mg, 收率85%)。
68.实施例37将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),2
‑
氯苯甲腈4k (137.6 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5k (168.0 mg, 收率70%)。
69.实施例38将反应容器中加入2
‑
氨基
‑5‑
氟苯甲醇1l (282.1 mg, 2.0 mmol),苯甲腈4a (103.1 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5l (134.4 mg, 收率60%)。
70.实施例39将反应容器中加入2
‑
氨基
‑5‑
氯苯甲醇1m (315.2 mg, 2.0 mmol),苯甲腈4a (103.1 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5m (153.6 mg, 收率64%)。
71.实施例40将反应容器中加入2
‑
氨基
‑5‑
溴苯甲醇1n (404.1 mg, 2.0 mmol),苯甲腈4a (103.1 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5n (207.3 mg, 收率73%)。
72.实施例41
将反应容器中加入2
‑
氨基
‑3‑
甲基苯甲醇1o (274.4 mg, 2.0 mmol),苯甲腈4a (103.1 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5o (169.5 mg, 收率77%)。
73.实施例42将反应容器中加入2
‑
氨基
‑3‑
甲基甲醇1o (274.4 mg, 2.0 mmol),4
‑
甲基苯甲腈4b (133.2 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5p (166.2 mg, 收率71%)。
74.实施例43将反应容器中加入2
‑
氨基
‑3‑
甲基苯甲醇1o (274.4 mg, 2.0 mmol),4
‑
溴苯甲腈4e (182.0 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5q (187.7 mg, 收率63%)。
75.对比例1将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),苯乙酮2a (120.2 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol),ko
t
bu (112.0 mg, 1.0 mmol)和邻菲啰啉(14.4 mg, 0.08 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3a (133.3 mg, 收率65%)。从此结果看,在体系中加入配体邻菲啰啉不仅不会提升收率,还会使产品收率大大降低。
76.对比例2将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),苯甲腈4a (103.1 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol),ko
t
bu (112.0 mg, 1.0 mmol)和邻菲啰啉(14.4 mg, 0.08 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5a (82.4 mg, 收率40%)。从此结果看,在体系中加入配体邻菲啰啉不仅不会提升收率,还会使产品收率大大降低。
77.对比例3将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),苯乙酮2a (120.2 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和cs2co
3 (325.0 mg, 1.0 mmol)。
在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3a (86.1 mg, 收率42%)。从此结果看,将碱换为cs2co
3 后收率大大降低。
78.对比例4将反应容器中加入2
‑
氨基苯甲醇1a (246.4 mg, 2.0 mmol),苯甲腈4a (103.1 mg, 1.0 mmol),co(oac)2·
4h2o(19.9 mg, 0.08 mmol)和k2co
3 (138.0 mg, 1.0 mmol)。在氩气气氛下,加入4 ml的叔戊醇并在95℃下搅拌24 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机层在减压下浓缩。然后将残余物通过快速柱色谱法(石油醚:乙酸乙酯100∶1, v / v)纯化,得到白色固体5a (127.7 mg, 收率62%)。从此结果看,将碱换为k2co3后收率大大降低。
79.对比例5将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),苯乙酮2a (120.2 mg, 1.0 mmol),cocl2(10.4 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到白色固体3a (112.8 mg, 收率55%)。从此结果看,将醋酸钴换为cocl2后收率大大降低。
80.对比例6将反应容器中加入2
‑
氨基苯甲醇1a (123.2 mg, 1.0 mmol),丙酮2v (58.1 mg, 1.0 mmol),co(oac)2·
4h2o (19.9 mg, 0.08 mmol)和ko
t
bu (112.0 mg, 1.0 mmol)。在氩气气氛下,加入2 ml甲苯并在110℃下加热12 h。冷却至室温后,添加10 ml水,并将混合物用etoac (3
×
10 ml)萃取。合并的有机相在减压下浓缩。然后将残余物通过快速柱色谱纯化(石油醚:乙酸乙酯100∶1, v / v),得到黄色油状液体3v (30.8 mg, 收率15%)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1