一种石墨烯增强呋喃基聚酯复合材料及其制备方法
1.本发明属于高分子复合材料技术领域,具体涉及一种石墨烯增强呋喃基聚酯复合材料及其制备方法。
背景技术:
2.传统聚合物单体一般来源于化石原料,消耗了大量的石油资源,同时造成了环境污染。随着可持续发展的观念深入人心,从生物质原料的转化中获得许多呋喃基单体,其具有可再生、原料来源广,且副产物少、收率高等优点,广泛应用于可持续化学的发展。聚酯因其良好的生物相容性、生物降解性被用于可降解塑料、药物包装及载体、绿色涂料、环保型热塑胶等领域,但由于其热性能和机械性能差,不适合采用一般的塑料成型工艺,需要对聚酯产品进行进一步的优化和改良。
3.石墨烯是一种由碳原子以sp2杂化轨道组成六角型呈蜂巢晶格的二维碳纳米材料,其可以从天然丰富、低成本的石墨中提取出来,并且因其具有优异的力学性能、导热性和导电性,非常适用于高性能聚合物纳米复合材料。由于聚合物本身结构性能的不足,可通过在聚酯结构中加入石墨烯,利用石墨烯上面的含氧基团制备得到呋喃基聚酯/石墨烯纳米复合材料,可增强聚合物本身的韧性,同时也可以提高聚酯材料的热稳定性,实现性能优异的新型结构的聚酯材料绿色可持续性生产。
技术实现要素:
4.发明目的:为解决现有技术中存在的技术问题,本发明提供一种石墨烯增强呋喃基聚酯复合材料及其制备方法,以解决现有技术中存在的依赖于石油基资源和新型聚酯材料缺少及其产品性能不佳等缺点。
5.为实现上述目的,本发明提出一种石墨烯增强呋喃基聚酯复合材料及其制备方法,包括以下步骤:
6.(a)将脂肪族环酯、引发剂和第一催化剂在溶剂中混合后,进行开环聚合反应,得到双羟基封端的聚酯预聚体;
7.(b)在氧化石墨烯存在下,第二催化剂催化聚酯预聚体和呋喃基单体缩聚反应,原位聚合获得呋喃基聚酯/石墨烯纳米复合材料。
8.优选地,步骤(a)中的脂肪族环酯为戊内酯、己内酯和三亚甲基碳酸酯中的任意一种。申请人尝试过丙交酯,但是聚丙交酯预聚体与呋喃单体缩聚效果不好,性能差。
9.步骤(a)中的引发剂为二元醇或二元胺。
10.所述二元醇为1,4-丁二醇、1,6-己二醇、1,8-辛二醇、1,10-癸二醇、对苯二甲醇中的任意一种;所述二元胺为1,4-丁二胺、1,5-戊二胺、1,6-己二胺,1,8-辛二胺、对苯二甲胺中的任意一种。
11.步骤(a)中的第一催化剂为辛酸亚锡、1,5,7-三氮杂二环、1,8-二氮杂二环十一碳-7-烯、磷酸二苯酯中的任意一种。
12.步骤(a)中有机溶剂为甲苯、二苯基醚、二甲苯、二氯甲烷中的任意一种。有机溶剂的量根据单体摩尔比进行调整,保证相同的单体浓度即可。
13.步骤(a)中脂肪族环酯:引发剂:催化剂摩尔比为(10-50):1:(0.1%-1.0%)。
14.优选地,步骤(b)中呋喃基单体包括2,5-呋喃二甲酸、2,5-呋喃二甲酸二甲酯和2,5-呋喃二甲酰氯中的任意一种。上述原料来源广泛,其中2,5-呋喃二甲酸二甲酯可改善产物着色且性能优异。
15.步骤(b)中所用的第二催化剂为钛酸四丁酯、辛酸亚锡、三氧化二锑中的一种,催化剂用量为相对于聚酯预聚物的质量的0.1-2.0wt%;聚酯预聚体、呋喃基单体的投料摩尔比为1.0:(1.0-2.0),反应温度为140℃-240℃,优选180℃-220℃,温度升高有利于正向反应,但温度过高会影响产物着色,反应时间2.0h-5.0h。
16.步骤(b)中氧化石墨烯为单层或者厚度为0.5-3.0nm的多层结构,纯度99%,用量相对于聚酯预聚物的质量比为0.1-1.0wt%。石墨烯用量过低导致原位聚合效果不明显,过高发生团聚导致性能差。
17.有益效果:与现有技术相比,本发明具有如下优点:
18.(1)本发明利用有机催化和金属催化技术结合制备含石墨烯结构的新型呋喃基聚酯复合材料,实现了聚酯复合材料的可持续生产;
19.(2)本发明通过原位聚合过程中聚酯预聚体、呋喃基单体与氧化石墨烯上面的含氧基团之间发生反应,实现了性能更加优异的聚酯复合材料的高效合成;
20.(3)本发明合成的呋喃基聚酯/石墨烯纳米复合结构原料绿色,结构更加新颖,性能更加优异,具有绿色,安全,高效,应用范围广等优点。
附图说明
21.图1为实施例1聚戊内酯预聚体1h nmr图;
22.图2为呋喃基聚戊内酯复合前后的红外光谱图,其中,go为单纯氧化石墨烯的红外光谱,pvlf为呋喃基聚戊内酯的红外光谱图,pvlf-go为呋喃基聚戊内酯/石墨烯纳米复合材料的红外光谱图;
23.图3为呋喃基聚戊内酯复合前后的tga图,其中,pvl为聚戊内酯预聚体的tga图,pvlf为呋喃基聚戊内酯的tga图,pvlf-go为呋喃基聚戊内酯/石墨烯纳米复合材料的tga图;
24.图4为呋喃基聚碳酸酯复合前后的红外光谱图,其中,go为单纯氧化石墨烯的红外光谱,ptmcf为呋喃基聚碳酸酯的红外光谱图,ptmcf-go为呋喃基聚碳酸酯/石墨烯纳米复合材料的红外光谱图;
25.图5为呋喃基聚碳酸酯复合前后的tga图,其中,ptmc为聚碳酸酯预聚体的tga图,ptmcf为呋喃基聚碳酸酯的tga图,ptmcf-go为呋喃基聚碳酸酯/石墨烯纳米复合材料的tga图;
26.图6为实施例8聚碳酸酯预聚体的1h nmr图;
27.图7为实施例9聚己内酯预聚体的1h nmr图;
28.图8为呋喃基聚己内酯复合前后的红外光谱图,其中,go为单纯氧化石墨烯的红外光谱,pclf为呋喃基聚己内酯的红外光谱图,pclf-go为呋喃基聚己内酯/石墨烯纳米复合
材料的红外光谱图;
29.图9为呋喃基聚己内酯复合前后的tga图,其中,pcl为聚己内酯预聚体的tga图,pclf为呋喃基聚己内酯的tga图,pclf-go为呋喃基聚己内酯/石墨烯纳米复合材料的tga图;
30.图10为实施例9含呋喃基聚己内酯/石墨烯纳米复合材料合成路线。
具体实施方式
31.下面结合具体实施例对本发明做进一步详细说明,实施例将有助于理解本发明,但是本发明的保护范围不限于下述的实施例。
32.本发明的下述实施例中,采用400mhz的bruker核磁共振仪器对产物的分子量进行测量:取聚酯预聚体样品10mg与核磁管中,加入氘代氯仿,待完全溶解后测样。
33.本发明的下述实施例中,采用渗透凝胶色谱法(gpc)对产物的分子量及分子量分布,以四氢呋喃(thf)为溶剂,0.7ml/min的流速进行测量,分子量以聚苯乙烯标样校准。
34.本发明的下述实施例中,采用bruker的红外光谱仪(分辨率为2cm-1
),用kbr压片后进行表征,波长范围400-4000cm-1
。
35.本发明的下述实施例中,采用tga550的热重分析仪对产物的热学性能进行测量:温度范围为30℃-800℃,升温速率为10℃,质量损失为5.0%的时候为初始分解温度。
36.本发明的下述实施例中,拉伸和弯曲性能通过zwick z020万能材料试验机测得,拉伸性能按照gb1040-2测得,根据gb/t 1040.2-2006方法测得呋喃基聚酯的韧性。
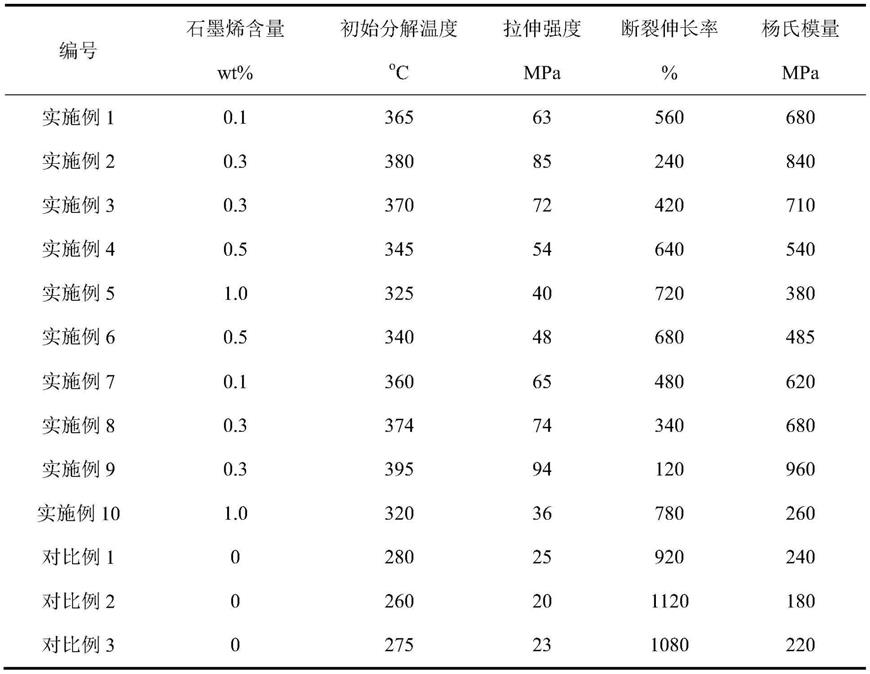
37.实施例1
38.在惰性气体保护下,分别将戊内酯(50mmol,5.0060g),引发剂丁二醇(5mmol,0.4506g),1,8-二氮杂二环十一碳-7-烯(0.25mmol,0.0381g)和20ml二甲苯溶剂置于反应瓶中,在25℃下反应0.5h后,滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚戊内酯产物,核磁图谱如图1所示,经核磁分析转化率为96%,通过1h nmr得到数均分子量为1046g/mol,pdi为1.06,初始分解温度t
d,5%
为206℃。
39.将聚戊内酯(2.8680mmol,3.0g),2,5-呋喃二甲酸二甲酯(2.8680mmol,0.5281g),氧化石墨烯(0.0030g,0.1wt%),钛酸四丁酯(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至220℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚戊内酯/石墨烯纳米复合材料。图2为制备的呋喃基聚戊内酯/石墨烯纳米复合材料红外光谱图,表明两者化学结合。产率为78%,图3为呋喃基聚戊内酯/石墨烯纳米复合材料的tga图,初始分解温度t
d,5%
为365℃,拉伸强度为63mpa,断裂伸长率为560%,杨氏模量680mpa。
40.实施例2
41.在惰性气体保护下,分别将己内酯(50mmol,5.7070g),引发剂己二醇(2mmol,0.2363g),磷酸二苯酯(0.25mmol,0.0626g)和19.6ml甲苯溶剂置于反应瓶中,在25℃下反应2.0h后,滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤
三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚己内酯产物,经核磁分析转化率为95%,通过1h nmr得到数均分子量为2710g/mol,pdi为1.10,初始分解温度t
d,5%
为235℃。
42.将聚己内酯(1.1070mmol,3.0g),2,5-呋喃二甲酸二甲酯(1.6605mmol,0.3058g),氧化石墨烯(0.0090g,0.3wt%),钛酸四丁酯(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至200℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚己内酯/石墨烯纳米复合材料。产率为82%,初始分解温度t
d,5%
为380℃,拉伸强度为85mpa,断裂伸长率为240%,杨氏模量840mpa。
43.实施例3
44.在惰性气体保护下,分别将三亚甲基碳酸酯(50mmol,5.1045g),引发剂辛二醇(5mmol,0.7312g),1,5,7-三氮杂二环(0.50mmol,0.0696g)和19.8ml二氯甲烷溶剂置于反应瓶中,在25℃下反应2.0h后,滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚碳酸酯产物,经核磁分析转化率为92%,通过1h nmr得到数均分子量为910g/mol,pdi为1.06,初始分解温度t
d,5%
为215℃。
45.将聚碳酸酯(3.2967mmol,3.0g),2,5-呋喃二甲酸(3.2967mmol,0.5146g),氧化石墨烯(0.0090g,0.3wt%),钛酸四丁酯(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至200℃,将反应体系的压力由大气压缓慢降至100pa以下反应3.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚碳酸酯/石墨烯纳米复合材料,产率为70%,初始分解温度t
d,5%
为370℃,拉伸强度为72mpa,断裂伸长率为420%,杨氏模量710mpa。
46.实施例4
47.在惰性气体保护下,分别将己内酯(50mmol,5.7070g),引发剂癸二醇(5mmol,0.8714g),辛酸亚锡(0.05mmol,0.0203g)和19.6ml二苯基醚溶剂置于反应瓶中,在140℃下反应2.0h后,冷却后滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚己内酯产物,经核磁分析转化率为90%,通过1h nmr得到数均分子量为1010g/mol,pdi为1.08,初始分解温度t
d,5%
为220℃。
48.将聚己内酯(2.9703mmol,3.0g),2,5-呋喃二甲酸二甲酯(2.9703mmol,0.5470g),氧化石墨烯(0.0150g,0.5wt%),三氧化二锑(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至220℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚己内酯/石墨烯纳米复合材料。产率为65%,初始分解温度t
d,5%
为345℃,拉伸强度为54mpa,断裂伸长率为640%,杨氏模量540mpa。
49.实施例5
50.在惰性气体保护下,分别将己内酯(50mmol,5.7070g),引发剂对苯二醇(1mmol,
0.1382g),1,5,7-三氮杂二环(0.50mmol,0.0696g)和19.6ml甲苯溶剂置于反应瓶中,在25℃下反应2.0h后,滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚己内酯产物,经核磁分析转化率为98%,通过1h nmr得到数均分子量为5490g/mol,pdi为1.12,初始分解温度t
d,5%
为245℃。
51.将聚己内酯(0.5464mmol,3.0g),2,5-呋喃二甲酰氯(1.0928mmol,0.2109g),氧化石墨烯(0.0300g,1.0wt%),辛酸亚锡(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至240℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚己内酯/石墨烯纳米复合材料。产率为60%,初始分解温度t
d,5%
为325℃,拉伸强度为40mpa,断裂伸长率为720%,杨氏模量380mpa。
52.实施例6
53.在惰性气体保护下,分别将戊内酯(50mmol,5.0060g),引发剂丁二胺(1mmol,0.0882g),1,8-二氮杂二环十一碳-7-烯(0.50mmol,0.0761g)和20.0ml二甲苯溶剂置于反应瓶中,在25℃下反应2.0h后,滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚戊内酯产物,经核磁分析转化率为92%,通过1h nmr得到数均分子量为4510g/mol,pdi为1.12,初始分解温度t
d,5%
为235℃。
54.将聚戊内酯(0.6652mmol,3.0g),2,5-呋喃二甲酸(1.3304mmol,0.2077g),氧化石墨烯(0.0150g,0.5wt%),钛酸四丁酯(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至200℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚己内酯/石墨烯纳米复合材料。产率为72%,初始分解温度t
d,5%
为340℃,拉伸强度为48mpa,断裂伸长率为680%,杨氏模量485mpa。
55.实施例7
56.在惰性气体保护下,分别将戊内酯(50mmol,5.0060g),引发剂对苯二胺(2mmol,0.2724g),1,5,7-三氮杂二环(0.25mmol,0.0348g)和20.0ml甲苯溶剂置于反应瓶中,在25℃下反应2.0h后,滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚戊内酯产物,经核磁分析转化率为96%,通过1h nmr得到数均分子量为2640g/mol,pdi为1.10,初始分解温度t
d,5%
为228℃。
57.将聚戊内酯(1.1364mmol,3.0g),2,5-呋喃二甲酰氯(1.1364mmol,0.2193g),氧化石墨烯(0.0030g,0.1wt%),辛酸亚锡(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至240℃,将反应体系的压力由大气压缓慢降至100pa以下反应3.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚戊内酯/石墨烯纳米复合材料。产率为75%,初始分解温度t
d,5%
为360℃,拉伸强度
为65mpa,断裂伸长率为480%,杨氏模量620mpa。
58.实施例8
59.在惰性气体保护下,分别将三亚基碳酸酯(50mmol,5.1045g),引发剂辛二胺(5mmol,0.7314g),辛酸亚锡(0.05mmol,0.0203g)和20.0ml二苯基醚溶剂置于反应瓶中,在140℃下反应2.0h后,滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚碳酸酯产物,图6为聚碳酸酯预聚体1h nmr图,经核磁分析转化率为90%,通过1h nmr得到数均分子量为920g/mol,pdi为1.08,初始分解温度t
d,5%
为220℃。
60.将聚碳酸酯(3.2609mmol,3.0g),2,5-呋喃二甲酸二甲酯(4.8913mmol,0.9007g),氧化石墨烯(0.0090g,0.3wt%),三氧化二锑(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至220℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚碳酸酯/石墨烯纳米复合材料。产率为76%,图4为呋喃基聚碳酸酯/石墨烯纳米复合材料的红外光谱图,图5为呋喃基聚碳酸酯/石墨烯纳米复合材料tga图,初始分解温度t
d,5%
为374℃,拉伸强度为74mpa,断裂伸长率为340%,杨氏模量680mpa。
61.实施例9
62.图10为实施例9含呋喃基聚己内酯/石墨烯纳米复合材料合成路线。在惰性气体保护下,分别将己内酯(50mmol,5.7070g),引发剂戊二胺(5mmol,0.5109g),1,5,7-三氮杂二环(0.25mmol,0.0348g)和19.6ml甲苯溶剂置于反应瓶中,在25℃下反应1.0h后,滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚己内酯产物,图7为聚己内酯预聚体1h nmr图,经核磁分析转化率为98%,通过1h nmr得到数均分子量为1220g/mol,pdi为1.10,初始分解温度t
d,5%
为230℃。
63.将聚己内酯(2.4590mmol,3.0g),2,5-呋喃二甲酸二甲酯(3.6885mmol,0.6792g),氧化石墨烯(0.0090g,0.3wt%),钛酸四丁酯(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至200℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚己内酯/石墨烯纳米复合材料。产率为82%,图8为呋喃基聚己内酯/石墨烯纳米复合材料红外光谱图,图9为呋喃基聚己内酯/石墨烯纳米复合材料tga图,初始分解温度t
d,5%
为395℃,拉伸强度为94mpa,断裂伸长率为120%,杨氏模量960mpa。
64.实施例10
65.在惰性气体保护下,分别将己内酯(50mmol,5.7070g),引发剂己二胺(2mmol,0.2324g),1,5,7-三氮杂二环(0.50mmol,0.0696g)和19.6ml甲苯溶剂置于反应瓶中,在25℃下反应2.0h后,滴加入冷甲醇溶液中进行沉淀,离心,去除上清液,反复加入二氯甲烷溶液洗涤三次,置于真空干燥箱中35℃下干燥24h,得到纯净的聚己内酯产物,经核磁分析转化率为96%,通过1h nmr得到数均分子量为2810g/mol,pdi为1.12,初始分解温度t
d,5%
为238℃。
66.将聚己内酯(1.0676mmol,3.0g),2,5-呋喃二甲酸(1.0676mmol,0.1666g),氧化石墨烯(0.0300g,1.0wt%),钛酸四丁酯(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下氮气保护反应2.0h,接着升温至200℃,将反应体系的压力由大气压缓慢降至100pa以下反应3.0h进行原位聚合反应,冷却后用二氯甲烷溶解,过滤除去过量石墨烯,将滤液滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到纯净的呋喃基聚己内酯/石墨烯纳米复合材料。产率为65%,初始分解温度t
d,5%
为320℃,拉伸强度为36mpa,断裂伸长率为780%,杨氏模量260mpa。
67.对比例1
68.将数均分子量为1046g/mol的聚戊内酯(2.8680mmol,3.0g),2,5-呋喃二甲酸二甲酯(2.8680mmol,0.5281g),钛酸四丁酯(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下惰性气体保护下反应2.0h,接着升温至200℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行缩聚反应,冷却后用二氯甲烷溶解,滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到呋喃基聚戊内酯,其红外光谱图如图2所示,tga图如图3所示,产率为80%,初始分解温度t
d,5%
为280℃,拉伸强度为25mpa,断裂伸长率为920%,杨氏模量240mpa。
69.对比例2
70.将数均分子量为910g/mol的聚碳酸酯(3.2967mmol,3.0g),2,5-呋喃二甲酸二甲酯(3.2967mmol,0.6070g),钛酸四丁酯(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下惰性气体保护下反应2.0h,接着升温至200℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行缩聚反应,冷却后用二氯甲烷溶解,滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到呋喃基聚碳酸酯,产率为70%,红外光谱图如图4所示,tga图如图5所示,初始分解温度t
d,5%
为260℃,拉伸强度为20mpa,断裂伸长率为1120%,杨氏模量180mpa。
71.对比例3
72.将数均分子量为1010g/mol的聚己内酯(2.9703mmol,3.0g),2,5-呋喃二甲酸二甲酯(2.9703mmol,0.5470g),钛酸四丁酯(0.0600g,2wt%)加入到惰性气体保护的反应瓶中。加热至140℃下惰性气体保护下反应2.0h,接着升温至200℃,将反应体系的压力由大气压缓慢降至100pa以下反应5.0h进行缩聚反应,冷却后用二氯甲烷溶解,滴加入冷甲醇中沉淀,离心洗涤三次,置于35℃真空干燥箱中干燥24h,得到呋喃基聚己内酯,产率为75%,红外光谱图如图8所示,tga图如图9所示,初始分解温度t
d,5%
为275℃,拉伸强度为23mpa,断裂伸长率为1080%,杨氏模量220mpa。
73.表1石墨烯/呋喃基聚酯制备与性能表征
[0074][0075]
本发明提供了一种石墨烯增强呋喃基聚酯复合材料及其制备方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1