一种苯并异喹啉的制备方法与流程
1.本发明涉及一种苯并异喹啉的制备方法,具体涉及的是盐酸帕洛诺司琼杂质的制备方法。
背景技术:
2.盐酸帕洛诺司琼对5-ht3受体有高选择性拮抗作用,可阻断呕吐反射中枢外周神经元的突触前5-ht3受体的兴奋,并且直接影响中枢神经系统的内5-ht3受体传递的迷走神经传入后区的作用,阻断肠道中迷走神经末梢,阻止信号传递到5-ht3受体触发区,减少呕吐和恶心的发生率。
3.盐酸帕洛诺司琼的结构式如下所示:
[0004][0005]
文献1(journal of medicinal chemistry,1993,vol.36,i18,2645-2657)描述了一种由5,6,7,8-四氢-1-萘甲酸合成n-[(s)-1-氮杂双环[2.2.2]辛-3-基]-5,6,7,8-四氢萘-1-甲酰胺,再得到2-[(s)-1-氮杂双环[2.2.2]辛-3-基]-2,4,5,6-四氢-1h-苯并[de]异喹啉-1-酮盐酸盐的方法。其化学反应式如下:
[0006][0007]
该方法使用到氯化亚砜,正丁基锂,且反应需在-22℃~-14℃下进行。专利us5202333也描述了类似的方法。
[0008]
文献2(synthesis,2000,i8,1113-1116)描述了一种由5,6,7,8-四氢-1-萘甲酸合成5,6-二氢-1h,4h-萘[1,8-cd]吡喃-1-酮,再得到2-[(s)-1-氮杂双环[2.2.2]辛-3-基]-2,4,5,6-四氢-1h-苯并[de]异喹啉-1-酮的方法,其化学反应式如下:
[0009][0010]
该方法同样使用到氯化亚砜和正丁基锂,且步骤更加繁琐。文献3(acs chemical neuroscience,2016,vol7,i11,1552-1564)也描述了类似的方法。
[0011]
在反应过程中,2,3-位双键会发生迁移形成3,4-位双键杂质,对盐酸帕洛诺司琼的质量造成不利影响。因此研究该杂质,提供控制策略,提高盐酸帕洛诺司琼的质量水平是合成盐酸帕洛诺司琼的重要工作之一。
技术实现要素:
[0012]
本发明的目的之一,在于提供一种式ⅰ化合物的制备方法,该方法为研究盐酸帕洛诺司琼杂质提供了一种新思路。
[0013]
为了实现上述目的,本发明采用以下技术方案:
[0014]
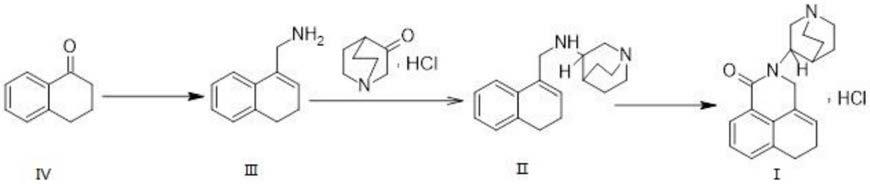
基于一种式ⅰ化合物的制备方法,具体包括以下步骤:
[0015]
将式ⅱ化合物与化合物c进行环合反应得到式ⅰ化合物。
[0016][0017]
进一步,所述化合物c为三光气。
[0018]
进一步,所述环合反应在溶剂5中反应,反应温度为0~40℃;所述溶剂5为甲苯、乙苯和二甲苯中任意一种。
[0019]
本发明的目的之二,在于提供一种式ⅱ化合物的制备方法,该方法为研究盐酸帕洛诺司琼杂质提供了一种新思路。
[0020]
为了实现上述目的,本发明采用以下技术方案:
[0021]
基于一种式ⅱ化合物的制备方法,具体包括以下步骤:
[0022]
1)将式ⅲ化合物与化合物b在碱作用下进行取代反应2;
[0023]
2)在催化剂b作用下经还原反应2得到式ⅱ化合物。
[0024]
[0025]
进一步,所述化合物b为奎宁酮盐。
[0026]
进一步,所述取代反应2在溶剂3中与碱反应,反应温度为40~80℃。
[0027]
进一步,所述溶剂3为甲苯、乙苯和二甲苯中任意一种;所述碱为碳酸钠、碳酸氢钠、三乙胺、二乙胺和吡啶中任意一种。
[0028]
进一步,所述还原反应2在溶剂4中反应,反应温度为10~40℃;所述溶剂4为甲醇、乙醇、异丙醇和正丁醇中任意一种。
[0029]
本发明的目的之三,在于提供一种式ⅲ化合物的制备方法,该方法为研究盐酸帕洛诺司琼杂质提供了一种新思路。
[0030]
为了实现上述目的,本发明采用以下技术方案:
[0031]
基于一种式ⅲ化合物的制备方法,具体包括以下步骤:
[0032]
1)以式ⅳ化合物与化合物a进行取代反应1;
[0033]
2)在催化剂a作用下进行还原反应1;
[0034]
3)经酸消除反应得到式ⅲ化合物。
[0035][0036]
进一步,所述ⅳ化合物为α-四氢萘酮;所述化合物a为氰基硅烷。
[0037]
进一步,所述取代反应1采用路易斯酸催化,反应温度为20~60℃;所述路易斯酸为三氯化铝、溴化铁、溴化铝、氯化铜、氯化亚铜、溴化亚铜、碘化亚铜、四氯化锡和四氯化钛中任意一种。
[0038]
进一步,所述还原反应1在溶剂1中进行,反应温度为10~30℃;所述溶剂1为四氢呋喃、1,4-二氧六环和环氧己烷中任意一种。
[0039]
进一步,所述消除反应在溶剂2中进行,反应温度为40~80℃;所述溶剂2为甲醇、乙醇、异丙醇和正丁醇中任意一种。
[0040]
进一步,所述的苯并异喹啉化合物由以上制备方法中的一种、两种或三种制备而成。
[0041]
本发明的目的之四,在于提供一种由上述式ⅱ化合物、式ⅲ化合物和式ⅳ化合物制备的式ⅰ化合物,即苯并异喹啉化合物的制备方法,该方法为研究盐酸帕洛诺司琼杂质提供了一种新思路。
[0042]
为了实现上述目的,本发明采用以下技术方案:
[0043]
基于一种苯并异喹啉化合物的制备方法,具体包括以下步骤:
[0044]
1)将式ⅳ化合物溶于化合物a中反应,得到式ⅲ化合物;
[0045]
2)式ⅲ化合物与化合物b反应得到式ⅱ化合物;
[0046]
3)式ⅱ化合物与化合物c反应得到式ⅰ化合物。
[0047][0048]
本发明的有益效果:
[0049]
1)本发明选用的有机溶剂根据ich指南,均为二类或三类有机溶剂,避免了一类有机溶剂的使用,降低了对人体的有害程度,也更加环保;
[0050]
2)本发明操作简便,无超高温或超低温等复杂操作,制备方法更加安全可靠;
[0051]
3)本发明步骤简短,仅需三步即可获得目标产物,收率较高。
具体实施方式
[0052]
下面将结合实施例对本发明的技术方案进行清楚、完整地描述。所描述的实施例仅为本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0053]
实施例1:3,4-二氢-1-萘甲胺(式ⅲ化合物)的制备
[0054][0055]
在氮气保护下,三口瓶内加入14.7gα-四氢萘酮、0.1g无水三氯化铝以及11.9g三甲基氰基硅烷,升温至30~40℃反应过夜,tlc监控反应完全;加入四氢呋喃,降温至0~5℃加入氢化铝锂,升温至20~25℃反应3小时,tlc监控反应完全,加入46g十水硫酸钠,搅拌1小时后,过滤;将滤液浓缩得到20.5g油状物。加入630ml无水乙醇,加入63ml盐酸,升温至55~65℃反应过夜,tlc监控反应完全,减压浓缩至干,加入无水乙醇100ml,减压浓缩至断流;浓缩物加入50ml二氯甲烷分散,加入250ml异丙醚后降温析晶0.5小时,抽滤,干燥得到15.9g式ⅲ化合物。
[0056]
实施例2:n-[(3,4-二氢萘-1-基)甲基]奎宁环-3-胺双盐酸盐(式ⅱ化合物)的制备
[0057]
[0058]
氮气保护下,取10g化合物1溶于200ml甲苯,加入10g奎宁酮盐酸盐、14g三乙胺,升温至90~110℃反应6小时,降温,浓缩除去甲苯;加入200ml甲醇,降温至0~5℃加入6g硼氢化钠;然后升温至20~30℃反应过夜,tlc监控反应基本完全,滴加30ml饮用水,滴毕,浓缩除去甲醇;加入200ml水和200ml乙酸乙酯,分液后,用乙酸乙酯100ml
×
2、50ml
×
2萃取;合并有机层,洗涤后,加入50g无水硫酸镁和2g活性炭脱色干燥;抽滤浓缩得到10.1g式ⅱ化合物。
[0059]
另取3.3g上述油状物,溶于70ml无水乙醇,加入2.2ml盐酸,室温搅拌1小时,浓缩除去乙醇,再用无水乙醇30ml
×
2带蒸至干;加入40ml乙酸乙酯分散,再加入40ml正己烷,冰浴降温析晶1小时,抽滤,干燥得到4.0g式ⅱ化合物,用于ms及nmr分析。
[0060]
ms:m+h
+
=269.3,2m+h
+
=537.6,m=268。
[0061]1h-nmr:(600mhz,d2o):δ=7.265-7.278(m,4h),6.415-6.434(t,1h),4.124-4.246(dd,2h),3.912-3.943(m,1h),3.785-3.833(t,1h),3.337-3.385(m,3h),3.246-3.304(m,2h),2.733-2.765(t,j=8hz,2h),2.622-2.639(dd,1h),2.276-2.316(dd,2h),2.107-2.126(m,4h)ppm。
[0062]
13
c-nmr:(150mhz,d2o):δ=136.903,135.925,131.070,128.351,128.280,126.828,126.828,121.879,51.808,48.861,47.240,46.252,45.995,26.708,22.564,21.656,21.135,16.161ppm。
[0063]
实施例3:2-(奎宁环-3-基)-2,3,5,6-四氢-1h-苯并[de]异喹啉-1-酮盐酸盐(式ⅰ化合物)的制备
[0064][0065]
将6.7g化合物2和67ml甲苯加入反应瓶中,搅拌,溶解,降温至0~10℃,滴加67ml甲苯与29.7g三光气配制的溶液,滴毕,升温至20~30℃反应过夜;次日,冰浴降温,滴加30g三氟化硼乙醚,滴毕,升温至5~20℃搅拌1小时后,升温至90~100℃反5小时,tlc检测反应完全,降温至0~10℃,滴加18g盐酸和47ml饮用水配制的溶液,滴毕,加入56g水,升温至70~90℃反应1小时,tlc检测反应完全后,降至室温;将沉淀物抽滤后,滤液用水50ml
×
2萃取,将萃取液与滤饼合并转入三口瓶内,加入70ml乙酸乙酯,控制温度10~20℃滴加由26g氢氧化钠和56g水配置的碱液,调节ph值9~10;分液后,水层用乙酸乙酯70ml
×
2萃取,合并有机层,洗涤干燥后,浓缩得到5.8g油状物。将油状物柱层析提纯(洗脱剂:三氯甲烷:甲醇(v/v)=50:1~10:1(加入几滴三乙胺)),收集含化合物3的洗脱液,浓缩后得到3.8g油状物。将油状物溶于88ml异丙醇,滴加2.3g盐酸,滴毕,升温至75~85℃回流1小时,减压浓缩至干;加入32ml异丙醇热溶至澄清,滴加75ml正己烷,升温至回流,再滴加22ml正己烷;滴毕,降温至0~10℃析晶0.5小时,抽滤淋洗后干燥,得到3.2g式ⅰ化合物。
[0066]
ms:m+h
+
=295.0,m=294。
[0067]1h-nmr:(600mhz,d2o):δ=7.254-7.269(m,1h),7.193-7.223(t,2h),7.133-7.148(d,j=9.0hz,1h),4.309-4.389(m,2h),4.225-4.263(d,1h),3.717-3.748(m,1h),3.450-3.540(m,2h),3.286-3.372(m,3h),2.809-2.843(t,2h),2.285-2.336(m,3h),2.022-2.085(m,3h),1.948(m,1h)ppm。
[0068]
13
c-nmr:(150mhz,d2o):δ=173.952,149.122,136.957,130.041,129.794,128.469,128.219,126.843,123.199,49.242,49.186,47.911,46.555,46.083,26.791,24.409,22.076,17.881,17.563ppm。
[0069]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1