一种抗真菌化合物与应用
1.本发明属于医药技术领域,具体涉及一种抗真菌化合物及其药学上可接受的盐类及其应用。
背景技术:
2.近年来,随着免疫力低下人群(接受抗生素、糖皮质激素、免疫抑制剂和体内导管置入治疗)不断扩大,原本不致病的真菌演变成深部真菌感染病原体,并引起血液、内脏器官和脑等部位发生严重感染,甚至成为免疫缺陷患者的主要死因之一。常见的深部真菌感染病原体有念珠菌、曲霉菌、隐球菌等。目前,一种被称为“超级真菌”的耳念珠菌,由于其对临床一线药物均产生严重的耐药性和极高的致死率而备受科学家的关注。随着全球人口的老年化、艾滋病和糖尿病的病人增多,深部真菌感染的致病率和致死率不断攀升,每年造成100万人死亡,对人类健康造成严重的威胁。
3.目前,临床上有四类药物用于治疗系统性真菌感染:多烯类(两性霉素b)、核酸类(5-氟胞嘧啶)和唑类(氟康唑、伊曲康唑等)。随着临床上抗真菌药物的广泛使用,真菌耐药性日益严重,因此,临床上迫切需求新型、抗耐药的抗真菌药物。
技术实现要素:
4.本发明的第一目的是提供一种抗真菌化合物。
5.本发明的再一目的是提供一种所述抗真菌化合物在制备抗真菌药物中的应用。
6.为了实现上述目的,本发明采用的技术方案如下:
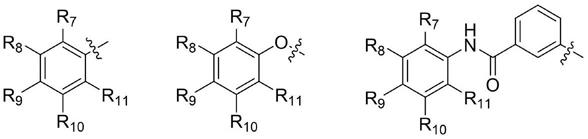
7.本发明的第一方面提供了一种抗真菌化合物或其药学上可接受的盐,具有以下结构通式:
[0008][0009]
x选自y选自-(ch2)
n-、n为0~3(优选0、1);
[0010]
r1选自
[0011][0012]
r7、r8、r9、r
10
、r
11
分别独立的选自氢、硝基、c1~c5烷基、c1~c5烷氧基、烯基取代的c1~c5烷氧基、炔基取代的c1~c5烷氧基、c3~c7环烷基取代的c1~c5烷氧基、乙烯基、乙炔基;
[0013]
r2、r3、r4、r5、r6分别独立的选自氢、c1~c5烷基、c1~c5烷氧基、烯基取代的c1~c5烷氧基、炔基取代的c1~c5烷氧基、c3~c7环烷基取代的c1~c5烷氧基、
[0014][0015]
或,r4、r5与c或一个n或两个n组成的六元环。
[0016]
较优选的,所述通式中,r7、r8、r9、r
10
、r
11
分别独立的选自氢、硝基、甲基、甲氧基、
[0017][0018]
r2、r3、r4、r5、r6分别独立的选自氢、甲基、甲氧基、
[0019][0020]
或,r4、r5与c或一个n或两个n组成的六元环为以下结构的一种:
[0021][0022]
更优选的,所述抗真菌化合物具有以下结构通式中的一种:
[0023][0024]
r2、r3、r4、r5、r6分别独立的选自氢、甲基、甲氧基、
[0025][0026]
r7、r8、r9、r
10
、r
11
分别独立的选自氢、硝基、甲基、甲氧基、
[0027][0028]
最优选的,所述抗真菌化合物选自以下结构的一种:
[0029]
[0030]
[0031][0032]
本发明的第二方面提供了一种所述抗真菌化合物或其药学上可接受的盐在制备抗真菌药物中的应用。
[0033]
所述真菌选自白念珠菌(c.albicans sc5314、c.albicans 103)、耳念珠菌(c.aur 0029、c.aur.0030、c.aur.15448)、新生隐球菌(c.neoformans h99)、热带念珠菌(c.tropical 10086)。
[0034]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0035]
本发明发现一类具有广谱抗真菌活性的苯甲酰胺类化合物及其类似物。部分化合物对白念珠菌、新生隐球菌、耐药白念珠菌、耐药热带念珠菌、耐药耳念珠菌表现出优秀的抗真菌活性。本发明优选部分化合物进行动物水平的抗真菌活性评价,结果表明,化合物b2和b7在耐药白念珠菌感染的体内模型中能显著降低小鼠的肾脏荷菌量,且化合物b7单独使用能够有效延长耐药真菌感染小鼠的生存期。在天然耐药的耳念珠菌感染模型中,化合物b2和b7能够显著降低小鼠肾脏耳念珠菌数量,表明化合物b2和b7经口服给药具有体内和体外抗耳念珠菌效果。
[0036]
本发明提供的化合物均为新型抗真菌小分子抑制剂,化合物中b2和b7在体外对多种真菌具有优秀的抗真菌活性,且在耐药菌感染模型中,化合物b2和b7对耐药白念珠菌103和耳念珠菌0029均具有体内抗真菌活性,因此本发明化合物可应用于制备抗真菌药物。
附图说明
[0037]
图1是化合物b2、b7的白念耐药菌感染小鼠肾脏荷菌量实验结果示意图。
[0038]
图2是化合物b2、b7的白念耐药菌感染小鼠生存期实验结果示意图。
[0039]
图3是化合物b2、b7的耳念珠菌感染小鼠肾脏荷菌量实验结果示意图。
具体实施方式
[0040]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0041]
下列实施例中所用试剂和原料均市售可得或可按文献方法制备。未注明具体条件的实验方法,按照常规条件,或按照制造厂商所建议的条件。
[0042]
1、化合物a系列的制备
[0043]
合成路线1
[0044][0045][0046]
试剂条件:(a)k2co3,ch3coch3,r.t,yield 89%;(b)fe,nh4cl,etoh,h2o,reflux,yield 93.6%;(c)hbtu,et3n,dmf,r.t,yield 35~62%;(d)pyridine,n2,dcm,reflux,
yield 39~41%;(e)ea,thf,h2o,na2co3,0℃,yield 26.7%.
[0047]
实施例1
[0048]
n-(3-烯丙基苯基)-4-甲氧基-3-硝基苯甲酰胺(a1)
[0049]
1-(烯丙氧基)-3-硝基苯(3a)
[0050]
间硝基苯酚(0.5g,3.5mmol)与烯丙基溴(0.64g,5.25mol)以及碳酸钾(0.97g,7mmol)溶于20ml丙酮溶剂中,室温条件下搅拌12h。tlc监测反应完成后,抽滤,3
×
20ml乙酸乙酯清洗滤饼,收集有机相,减压浓缩得淡黄色油状物0.57g,收率:89%。
[0051]
3-(烯丙氧基)苯胺(4a)
[0052]
化合物3a(0.57g,3.1mmol)和铁粉(0.95g,17.7mmol)溶于15ml乙醇,然后升温至50-55℃,加入氯化铵(0.52g,9.3mmol)和3ml水于85℃下回流反应2h。反应结束后过滤除去铁粉,30ml乙酸乙酯分两次清洗滤饼,回收滤液减压浓缩除去部分乙醇。将30ml水和20ml乙酸乙酯加入滤液,收集有机层,无水硫酸钠干燥,减压浓缩得0.44g黄色油状物0.44g,收率:93.6%。
[0053]
n-(3-烯丙基苯基)-4-甲氧基-3-硝基苯甲酰胺(a1)
[0054]
化合物4a(0.2g,1.34mmol)和-4-甲氧基-3-硝基苯甲酸(0.22g,1.24mmol)溶于n,n-二甲基甲酰胺于室温下搅拌,向其中加入缩合剂o-苯并三氮唑-四甲基脲六氟磷酸盐hbtu(0.56g,1.47mmol)和三乙胺(0.27g,2.68mmol)搅拌过夜。反应完成后,向反应液中加入35ml乙酸乙酯萃取,用100ml饱和食盐水洗4次,收集有机相,无水硫酸钠干燥,浓缩并经柱层析得0.16g白色固体即化合物a1,收率:40.3%。
[0055]
实施例2
[0056]
n1,n
3-双(3-(烯丙氧基)苯基)间苯二甲酰胺(a2)
[0057]
参考实施例1,化合物4a(0.18g,1.2mmol)与1,3-苯二甲酸(0.10g,0.6mmol)溶于n,n-二甲基甲酰胺于室温下搅拌,向其中加入缩合剂o-苯并三氮唑-四甲基脲六氟磷酸盐hbtu(0.56g,1.47mmol)和三乙胺(0.27g,2.68mmol)搅拌过夜。反应完成后,向反应液中加入35ml乙酸乙酯萃取,用100ml饱和食盐水洗4次,收集有机相,无水硫酸钠干燥,浓缩并经柱层析得到目标产物100mg,淡黄色固体,收率:38.9%。
[0058]
实施例3
[0059]
(e)-n-(3-(烯丙氧基)苯基)-3-(4-甲氧基苯基)丙烯酰胺(a3)
[0060]
参考实施例1,化合物4a与4-甲氧基肉桂酸反应得到目标产物90mg,白色固体,收率:35.1%。
[0061]
实施例4
[0062]
n-(3-(烯丙氧基)苯基)-4-乙炔基苯甲酰胺(a4)
[0063]
参考实施例1,化合物4a与4-乙炔基苯甲酸反应得到目标产物110mg,白色固体,收率:53%。
[0064]
实施例5
[0065]
n-(3-(烯丙氧基)苯基)-1h-吲唑-6-羧酰胺(a5)
[0066]
参考实施例1,化合物4a与1h-吲唑-6-羧酸反应得到目标产物136mg,白色固体,收率:47%。
[0067]
实施例6
[0068]
n-(3-(烯丙氧基)苯基)喹啉-3-羧酰胺(a6)
[0069]
参考实施例1,化合物4a与喹啉-3-羧酸反应得到目标产物75mg,白色固体,收率:42%。
[0070]
实施例7
[0071]
n-(3-(烯丙氧基)苯基)喹啉-7-羧酰胺(a7)
[0072]
参考实施例1,化合物4a与7-喹啉甲酸反应得到目标产物98mg,白色固体,收率:62%。
[0073]
实施例8
[0074]
n-(3-(烯丙氧基)苯基)-4-甲氧基苯磺酰胺(a8)
[0075]
称取化合物4a(80mg,0.528mmol)、4-甲氧基苯磺酰氯(0.1g,0.48mmol)和1ml吡啶溶于5ml的二氯甲烷中,氮气保护下室温反应2h。反应结束后,先用dcm(20ml)加以稀释,然后有机相依次用稀hcl、饱和nahco3和饱和nacl水溶液洗,最后收集有机相,无水na2so4干燥,减压浓缩并经柱层析得到62mg白色固体,收率为40.1%。
[0076]
实施例9
[0077]
n-(4-(烯丙氧基)苯基)喹啉-8-磺酰胺(a9)
[0078]
参考实施例8,化合物4a与8-喹啉磺酰氯反应得到目标产物101mg,白色固体,收率:39.5%。
[0079]
实施例10
[0080]
4-甲氧基苯基(3-(烯丙氧基)苯基)氨基甲酸酯(a10)
[0081]
称取化合物4a(0.14g,0.74mol)溶于乙酸乙酯、四氢呋喃和水的混合溶液中,于0℃条件下搅拌,然后加入化合物氯甲酸4-甲氧基苯酯(0.1g,0.67mmol)和碳酸氢钠(43mg,0.4mmol)反应1h,后移入室温条件下反应。反应结束后,先用etoac(30ml)萃取,接着用饱和nacl水溶液(2
×
20ml)洗涤,收集有机相,无水na2so4干燥,减压浓缩并经柱层析得到36mg黑色固体,产率:26.7%。
[0082]
2、化合物b系列的制备
[0083]
合成路线2
[0084][0085]
试剂条件:(a)hbtu,et3n,dmf,r.t,yield 39~82%.
[0086][0087]
实施例11
[0088]
n-(2-(烯丙氧基)苯基)喹啉-6-羧酰胺(b1)
[0089]
参考实施例1,化合物4b与6-喹啉甲酸反应得到目标产物170mg,黄色固体,收率:45.7%。
[0090]
实施例12
[0091]
n-(3-(烯丙氧基)苯基)喹啉-6-羧酰胺(b2)
[0092]
参考实施例1,化合物4a与6-喹啉甲酸反应得到目标产物89mg,淡黄色固体,收率:45.7%。
[0093]
实施例13
[0094]
n-(4-(烯丙氧基)苯基)喹啉-6-羧酰胺(b3)
[0095]
参考实施例1,化合物4c与6-喹啉甲酸反应得到目标产物120mg,白色固体,收率:62.1%。
[0096]
实施例14
[0097]
n-(3-(炔丙氧基)苯基)喹啉-6-羧酰胺(b4)
[0098]
参考实施例1,化合物4d与6-喹啉甲酸反应得到目标产物210mg,白色固体,收率:72%。
[0099]
实施例15
[0100]
n-(3-((2-(甲基氨基甲酰基)吡啶-4-基)氧基)苯基)喹啉-6-羧酰胺(b5)
[0101]
称取对氨基苯酚(1.28g,11.72mmol)和叔丁醇钾(1.62g,12.2mmol)溶解在dmf中,于室温下搅拌10分钟,然后加入碳酸钾(1.37g,12.2mmol)和n-甲基-4-氯-2-吡啶甲酰胺(2g,11.72mmol),于80℃下搅拌反应。反应结束后,先过滤,滤饼用etoac(2
×
20ml)洗涤,收集滤液接着连续用饱和nacl水溶液(4
×
20ml)洗,收集有机相,无水na2so4干燥,减压浓缩然后经柱层析得产物为1.12g,白色固体,产率39.7%。
[0102]
实施例16
[0103]
n-(2-(烯丙氧基)苯基)喹喔啉-6-羧酰胺(b6)
[0104]
参考实施例1,化合物4b与6-喹喔啉甲酸反应得到目标产物78mg,淡黄色固体,收率:39.6%。
[0105]
实施例17
[0106]
n-(3-(烯丙氧基)苯基)喹喔啉-6-羧酰胺(b7)
[0107]
参考实施例1,化合物4a与6-喹喔啉甲酸反应得到目标产物163mg,黄色固体,收率:57.4%。
[0108]
实施例18
[0109]
n-(4-(烯丙氧基)苯基)喹喔啉-6-羧酰胺(b8)
[0110]
参考实施例1,化合物4c与6-喹喔啉甲酸反应得到目标产物169mg,白色固体,收率:69.7%。
[0111]
实施例19
[0112]
n-(4-(炔丙氧基)苯基)喹喔啉-6-羧酰胺(b9)
[0113]
参考实施例1,化合物4d与6-喹喔啉甲酸反应得到目标产物155mg,白色固体,收率:58.8%。
[0114]
实施例20
[0115]
n-(3-((1-苄基-1h-1,2,3-三唑-4-基)甲氧基)苯基)喹啉-6-羧酰胺(b10)
[0116][0117]
(b)cu(oac)2,nan3,etoh,h2o,l-sodium ascorbate,1,10-phenanthroline,r.t,yield46.5%;
[0118]
称取乙酸铜(3mg,0.0165mmol)、1,10-菲罗啉(3mg,0.0165mmol)、l-抗血酸钠(65mg,0.33mmol),加入乙醇水的混合溶剂中,乙醇、水的体积比为4:1,搅拌5min,然后加入化合物b4(100mg,0.33mmol)、卞氯(46mg,0.363mmol)、叠氮化钠(24mg,0.363mmol),于室温下搅拌反应。反应结束后,加入etoac(3
×
20ml)萃取,饱和nacl水溶液(20ml)洗涤,收集有机相,无水na2so4干燥,减压浓缩经柱层析得目标产物67mg,白色固体,收率:46.5%。
[0119]
实施例21
[0120]
n-(3-((1-苄基-1h-1,2,3-三唑-4-基)甲氧基)苯基)喹啉-6-羧酰胺(b11)
[0121][0122]
(c)nh4cl,koh,meoh,r.t;yield 10.5%;
[0123]
参考实施例1,化合物3-(3-氨基苯氧基)丙酸甲酯和6-喹啉甲酸反应得到化合物7200mg,白色固体,收率;48.1%。
[0124]
称取化合物氯化铵(79mg,1.14mmol)和化合物7(200mg,0.57mmol)溶于甲醇中,然后缓慢加入氢氧化钾(64mg,1.14mmol),在常温条件下反应1h。反应结束后,反应液呈白色悬浮状,减压抽滤,收集滤液,用乙酸调ph为酸性,有固体析出,抽滤并用水洗涤,得目标产物(b11)21mg,白色固体,收率:10.5%。
[0125]
实施例22
[0126]
(s)-n-(3-(2-(2-(2-(羟甲基)吡咯烷基-1-基)乙氧基)苯基)喹啉-6-羧酰胺(b12)
[0127][0128]
(d)k2co3,dmf,60℃,yield 47.3%;
[0129]
参考实施例1,3-(2-溴乙氧基)苯胺和6-喹啉甲酸反应得到98mg化合物8(n-(3-(3-(羟氨基)-3-氧丙氧基)苯基)喹啉-6-羧酰胺),白色固体,收率;42.6%。
[0130]
称取化合物8(70mg,0.19mmol)和(s)-(+)-2-吡咯烷甲醇(20mg,0.19mmol)、碳酸
钾(52mg,0.38mmol)溶解在dmf中,于56℃下反应。反应结束后,加入etoac(30ml)萃取,饱和nacl水溶液(3
×
20ml)洗,收集有机相,无水na2so4干燥,减压浓缩并经柱层析得目标产物(b12)35mg,白色固体,收率:47.3%。
[0131]
实施例23
[0132]
n-(3-(2-羟基乙氧基)苯基)喹啉-6-甲酰胺(b13)
[0133][0134]
(e)ppts,ch3oh,r.t,yield40.1%.
[0135]
参考实施例1,3-(2-((四氢-2h-吡喃-2-基)氧基)乙氧基)苯胺和6-喹啉甲酸反应得到460mg化合物10(n-(3-(2-((四氢-2h-吡喃-2-基)氧基)乙氧基)苯基)喹啉-6-羧酰胺),黄色固体,收率:84.1%。
[0136]
称取化合物10(200mg,0.51mmol)和4-甲基苯磺酸吡啶(30mg,0.12mmol)溶解在甲醇溶液中,于室温条件下搅拌。反应结束后,先减压浓缩部分甲醇溶液,然后加入水(30ml)稀释浓缩液,并用etoac(2
×
25ml)进行萃取,收集有机相,无水na2so4干燥,减压浓缩并经柱层析得63mg目标产物b13,白色固体,收率:40.1%。
[0137]
合成路线3
[0138][0139]
试剂条件:(a)imidazole,dmf,r.t,yield 93%;(b)h2,pd/c,etoh,r.t,yield 83%;(c)hbtu,et3n,dmf,r.t,yield 56-73%;(d)lioh,dmf,r.t,yield 82-84%;(e)k2co3,etoh,reflux,yield 45-62%。
[0140]
实施例24
[0141]
n-(3-(环丙基甲氧基)苯基)喹啉-6-羧酰胺(b14)
[0142]
叔丁基二甲基(3-硝基苯氧基)硅烷(12)
[0143]
称取间硝基苯酚(1g,7.1mmol)、咪唑(725mg,10.65mmol)和三甲基氯硅烷(1.28g,8.52mmol)溶解在dmf中,于室温下搅拌。反应结束后,加入2m稀盐酸调ph为酸性,用etoac(2
×
30ml)萃取,收集有机相,饱和nacl水溶液洗3次,无水na2so4干燥,减压浓缩得黄色固体1g,产率为55%,直接进行下一步。
[0144]
3-((叔丁基二甲基甲硅烷基)氧基)苯胺(13)
[0145]
称取化合物12(1g,3.95mmol)和10%钯碳(30mg)溶解在乙醇溶液中,并保持在氢气气氛中,于室温条件下反应。反应结束后,先过滤除去钯碳,etoac(2
×
30ml)清洗滤饼,收
集有机相,减压浓缩得黑色油状物0.84g,产物为95.5%,直接进行下一步。
[0146]
n-(3-((叔丁基二甲基甲硅烷基)氧基)苯基)喹啉-6-羧酰胺(14a)
[0147][0148]
制备方法同目标化合物a1,化合物13与6-喹啉甲酸反应得到灰色固体200mg,产率:45.8%。
[0149][0150]
称取化合物14a(200mg,0.53mmol)和氢氧化锂(68.5mg,1.6mmol)溶解在dmf中,于室温下搅拌。反应结束后,向反应液加入1m稀盐酸调ph为酸性,反应液析出黄色固体124mg,产率:89.2%,直接进行下一步反应。
[0151]
n-(3-(环丙基甲氧基)苯基)喹啉-6-羧酰胺(b14)
[0152]
称取化合物15a(124mg,0.47mmol)、溴甲基环丙烷(310mg,2.3mmol)和碳酸钾(394mg,2.85mmol)溶解在n,n-二甲基甲酰胺溶液中,于室温下搅拌。反应结束后,过滤除去碳酸钾,加入50ml etoac进行萃取,饱和食盐水洗三次,收集有机相,无水硫酸钠干燥,减压浓缩并经柱层析得目标产物20mg,白色固体,收率:13.4%。
[0153]
实施例25
[0154]
n-(3-(环丙基甲氧基)苯基)喹喔啉-6-羧酰胺(b15)
[0155]
n-(3-((叔丁基二甲基硅基)氧基)苯基)喹喔啉-6-羧酰胺(中间体14b)
[0156][0157]
制备方法同目标化合物a1,中间体13与喹喔啉-6-羧酸反应得到灰色固体200mg,产率:45.8%。
[0158]
n-(3-羟基苯基)喹啉-6-甲酰胺(中间体15b)
[0159][0160]
称取中间体14b(200mg,0.53mmol)和氢氧化锂(68.5mg,1.6mmol)溶解在dmf中,于室温下搅拌。反应结束后,向反应液加入1m稀盐酸调ph为酸性,反应液析出黄色固体得124mg化合物15b,产率:89.2%,直接进行下一步反应。
[0161]
参考实施例24,中间体15b与溴甲基环丙烷反应得目标产物(b15)30mg,白色固体,收率:21.6%。
[0162]
3、化合物c系列的制备
[0163]
合成路线4
[0164][0165]
试剂条件:(a)dry dcm,r.t,yield 98%;(b)hbr/ch3cooh,70℃,yield 73.6%;(c)k2co3,etoh,reflux,yield 37%.
[0166]
实施例26
[0167]
1-(3-(烯丙氧基)苯基)-3-(6-喹啉基)脲(c1)
[0168]
1-(3-甲氧基苯基)-3-(6-喹啉基)脲(19)
[0169]
称取6-氨基喹啉(0.2g,1.4mmol)溶于二氯甲烷中,向反应液中缓慢滴加3-甲氧苯基异氰酸酯(0.2g,1.4mmol)和二氯甲烷的混合溶液,反应液可见有白色固体析出,反应继续搅拌1h。反应结束后,将反应液浓缩后直接进行下一步。
[0170]
1-(3-羟苯基)-3-(6-喹啉基)脲(20)
[0171]
称取化合物19(0.4g,1.3mmol)溶于33%氢溴酸/醋酸溶液中,于70℃条件下搅拌反应。反应结束,向反应液中加入2m氢氧化钠溶液调ph为碱性,加入乙酸乙酯进行萃取,收集有机相,无水硫酸钠干燥,减压浓缩并经柱层析得黑色固体0.28g,收率:73.6%。
[0172]
1-(3-(烯丙氧基)苯基)-3-(6-喹啉基)脲(化合物c1)
[0173]
称取化合物20(0.1g,0.36mmol)、烯丙基溴(70mg,0.54mmol)和碳酸钾(99mg,0.72mmol)在乙醇溶剂中回流反应。反应结束后,抽滤除去碳酸钾,乙酸乙酯清洗滤饼,有机溶剂浓缩并经柱层析得目标产物(c1)42mg,白色固体,收率:37%。
[0174]
合成路线5
[0175][0176]
试剂条件:(a)socl2,meoh,r.t,yield 95%;(b)nh
2-nh2·
h2o,meoh,reflux,yield 63.2%;(c)ch3cooh,meoh,r.t,yield 72-75%.
[0177]
实施例27
[0178]
(e)-n
’‑
(3-(烯丙氧基)亚苄基)喹喔啉-6-碳酰肼(c2)
[0179]
喹喔啉-6-羧酸甲酯(22)
[0180]
称取6-喹喔啉甲酸(1g,5.74mmol)和1ml氯化亚砜加入10ml甲醇溶液中,回流条件下搅拌反应。反应结束后,向反应液中加入水和乙酸乙酯进行萃取,收集有机相,无水硫酸钠干燥,浓缩并经柱层析得黄色油状物1.02g,收率:95%。
[0181]
喹喔啉-6-碳酰肼(23)
[0182]
称取化合物22(300mg,1.6mmol)和水合肼(1.2g,24mmol),在甲醇溶液中,回流条件下搅拌反应。反应结束后,将反应液旋干,加入2m稀盐酸调ph为中性,乙酸乙酯进行萃取,收集有机相,无水硫酸钠干燥,浓缩并经柱层析得白色油状物190.5mg,收率:63.2%。
[0183]
(e)-n
’‑
(3-(烯丙氧基)亚苄基)喹喔啉-6-碳酰肼(c2)
[0184]
称取化合物23(50mg,0.265mmol)和3-(烯丙氧基)苯甲醛24a(55mg,0.29mmol)溶于甲醇中,并加入两滴醋酸溶液于常温条件下反应。反应结束后,向反应液中加入水和乙酸乙酯进行萃取,收集有机相,无水硫酸钠干燥,浓缩并经柱层析得目标产物63.8mg,白色固体,收率:72.3%。
[0185]
实施例28
[0186]
(e)-n
’‑
(4-(烯丙氧基)亚苄基)喹喔啉-6-碳酰肼(c3)
[0187]
参考实施例27,中间体23与4-(烯丙氧基)苯甲醛24b反应得目标产物65.7mg,白色固体,收率:74.5%。
[0188]
合成路线6
[0189][0190]
试剂条件:(a)k2co3,etoh,reflux,yield 38%;(b)dipea,thf,0℃,yield 65-78%;(c)k2co3,etoh,85℃,yield 77-86%.
[0191]
实施例29
[0192]
2-(3-(烯丙氧基)苯氧基)-n-(6-喹啉基)乙酰胺(c4)
[0193]
3-(烯丙氧基)苯酚的合成(26a)
[0194][0195]
称取1,3-苯二酚(0.5g,3.59mmol)和烯丙基溴(652mg,5.39mmol)、碳酸钾(992mg,7.18mmol)在丙酮溶剂中回流反应。反应结束后,先抽滤,滤饼用etoac(30ml)洗涤,收集有
机相,减压浓缩并经柱层析得淡红色油状物。
[0196]
2-氯-n-(6-喹啉基)乙酰胺(中间体28a)
[0197][0198]
称取6-氨基喹啉(1g,6.9mmol)和n,n-二异丙基乙胺(1.12g,8.6mmol)溶解在thf溶液中,然后向其中滴加氯乙酰氯(0.78g,6.9mmol)和四氢呋喃的混合溶液,反应保持在0℃下搅拌反应。反应结束后,先加入50ml水进行淬灭,etoac(3
×
20ml)萃取,收集有机相,无水na2so4干燥,浓缩并经柱层析得目得黄色固体。
[0199]
称取中间体26a(0.1g,0.45mmol)、中间体28a(0.1g,0.675mmol)和碳酸钾(187mg,1.35mmol)在乙醇溶剂中回流搅拌反应。反应结束后,先过滤,etoac(2
×
20ml)清洗滤饼,收集有机相,减压浓缩并经过柱得目标产物(c4)116.6mg,白色固体。
[0200]
实施例30
[0201]
2-(3-(环丙基甲氧基)苯氧基)-n-(6-喹啉基)乙酰胺(c5)
[0202]
3-(环丙基甲氧基)苯酚的合成(中间体26b)
[0203][0204]
称取1,3-苯二酚(0.5g,3.59mmol)和溴甲基环丙烷(722mg,5.39mmol)、碳酸钾(992mg,7.18mmol)在丙酮溶剂中回流反应。反应结束后,先抽滤,滤饼用etoac(30ml)洗涤,收集有机相,减压浓缩并经柱层析得淡红色油状物。
[0205]
制备方法同实施例29,26b与28a反应得目标产物(c5)154mg,黄色固体,收率:81.2%。
[0206]
实施例31
[0207]
2-(3-(烯丙氧基)苯氧基)-n-(6-喹喔啉基)乙酰胺(c6)
[0208]
2-氯-n-(6-喹喔啉基)乙酰胺(中间体28b)
[0209][0210]
称取6-氨基喹喔啉(1g,6.9mmol)和n,n-二异丙基乙胺(1.12g,8.6mmol)溶解在thf溶液中,然后向其中滴加氯乙酰氯(0.78g,6.9mmol)和四氢呋喃的混合溶液,反应保持在0℃下搅拌反应。反应结束后,先加入50ml水进行淬灭,etoac(3
×
20ml)萃取,收集有机相,无水na2so4干燥,浓缩并经柱层析得目得黄色固体。
[0211]
参考实施例29,26a与28b反应得到目标产物94mg,白色固体,收率:79.2%。
[0212]
实施例32
[0213]
2-(3-(环丙基甲氧基)苯氧基)-n-(6-喹喔啉基)乙酰胺(c7)
[0214]
参考实施例29,26b与28b反应得到目标产物87mg,黄色固体,收率:78.5%。
[0215]
表1本发明部分化合物的nmr数据
[0216]
[0217]
[0218]
[0219]
[0220]
[0221]
[0222][0223]
本发明制备的化合物的性能测试:
[0224]
1、真菌生长抑制试验
[0225]
使用上述制备的化合物a1~a10、b1~b15、c1~c7进行真菌生长抑制试验,菌株选用临床常见致病真菌:白念珠菌(c.albicans sc5314、c.albicans 103)、耳念珠菌(c.aur 0029、c.aur.0030、c.aur.15448)、新生隐球菌(c.neoformans h99)和热带念珠菌(c.tropical 10086),阳性对照药为氟康唑(flc)和卡泊芬净(caspofungin)。
[0226]
材料及仪器
[0227]
移液器,96孔板,血细胞计数板,涡旋仪,玻璃管,ep管,超净化工作台(苏州安泰空气技术有限公司),thz-82a台式恒温振荡器(上海跃进医疗器械厂)。
[0228]
培养基配制
[0229]
yepd培养液:酵母浸膏10g,蛋白胨20g,葡萄糖20g,加三蒸水900ml溶解,加入2mg/ml氯霉素水溶液50ml,三蒸水定容至1000ml,高压灭菌(121℃,15min),于4℃保存备用。
[0230]
rpmi1640液体培养液:rpmi1640(gibco brl)10g,nahco32.0g,吗啡啉丙磺酸
(mops)34.5g,naoh 2.7g,三蒸水定容至1000ml,抽滤灭菌,于4℃保存备用。
[0231]
sda培养基:蛋白胨10g,琼脂20g,葡萄糖40g,三蒸水定容至1000ml,高压灭菌(121℃,15min),于4℃保存备用。
[0232]
pbs缓冲溶液:nacl 8.0g,na2hpo412h2o 3.57g,kcl 0.2g,kh2po40.24 g,三蒸水定容至1000ml,高压灭菌(121℃,15min),室温保存备用。
[0233]
待测药物的配制
[0234]
待测药物用dmso溶解,配制成2mg/ml的溶液,置于-20℃冰箱储存备用。
[0235]
菌悬液的制备
[0236]
用接种圈从4℃保存的sda培养基上挑取单克隆的菌株(致病真菌白念珠菌、新生隐球菌、耳念珠菌和热带念珠菌),接种至1mlyepd培养液中,于30℃,200rpm振荡培养,活化16h,使真菌处于指数生长期后期。取该菌液至1.5ml ep管,离心(3000g,5min,4℃),吸弃上清液,用1ml pbs缓冲液吹打混匀,用pbs缓冲溶液吹洗2次,用1ml rpmi 1640培养液吹打混匀,用血细胞计数板计数,以rpmi 1640培养液调整菌液浓度至1
×
103cells/ml。
[0237]
加药及检测
[0238]
将配制好的菌悬液加入96孔板中,第1列加入200μl,2至11列加入100μl,第12列加入rpmi1640培养基做空白对照。每个待测药物(2mg/ml)取6.4μl(部分化合物取1.6μl)于第一列菌悬液中,作三复孔,倍半稀释10个浓度梯度,即终浓度为64μg/ml(16μg/ml)-0.125μg/ml(0.03μg/ml),第11列不加药,作阳性对照。放入35℃恒温培养箱中孵育48h(c.alb.sc5314、c.alb.103、c.aur.0029、c.aur.0030,c.tro.10086)、c.aur.15448;72h(c.neo.h99),用酶标仪测定od
630
。以阳性对照的od值为100%,最低抑菌浓度(mic
80
)为80%od值的最低化合物浓度,实验结果如表2和表3所示。
[0239]
表2化合物体外抗真菌活性测试(mic
80
,μg/ml)
[0240][0241]aabbreviations:c.alb.,candida albicans,白念珠菌;c.tro.,candida tropicalis,热带念珠菌;c.neo.,cryptococcus neoformans,新生隐球菌;flc,
fluconazole,氟康唑.
[0242]
表3部分化合物对耳念珠菌的抗真菌活性测试(mic
80
,μg/ml,48h)
[0243][0244]aabbreviations:c.aur.,candida auris,耳念珠菌
[0245]
由表2和表3可知,体外抗真菌活性测试结果显示,a类化合物中a7具有中等抗真菌活性,其中,对新生隐球菌表现出较好的抗真菌活性;b类化合物b2-b4、b7、b14-b15,c类化合物c4-c5对耐药念珠菌、耳念珠菌和新生隐球菌都表现出优秀的抗真菌活性,部分发明的化合物活性优于对照药。
[0246]
2、体内实验
[0247]
本发明从上述制备的化合物中选取b2、b7进行小鼠体内抗真菌活性测试。测试方法包括:小鼠肾脏荷菌量实验和小鼠生存期实验。
[0248]
动物模型:选择icr雌性小鼠(20g左右)作为实验小鼠,每只小鼠经尾静脉注射3
×
105cfu/ml(0.2ml)的白念耐药菌和耳念珠菌的菌悬液建立小鼠系统性真菌感染模型。
[0249]
小鼠肾脏荷菌量实验:将小鼠分为10组,每组3只。空白对照为生理盐水组,给药方式分为口服给药和腹腔注射给药。
[0250]
每天给药一次,给药5天,第六天将小鼠处死,摘取肾脏、称重,将肾脏组织匀浆,经生理盐水稀释后,吸取20μl,均匀涂布在sda培养基上,每只小鼠匀浆液平行涂布3次。经35℃培养1.5天,计数培养基上菌落,计算并统计分析小鼠肾脏真菌荷菌量。
[0251]
小鼠生存期实验:将小鼠分为4组,每组10只。空白对照为生理盐水组,给药方式为腹腔注射。每天给药一次,共给药7天,连续记录各组小鼠生存时间,并统计分析。
[0252]
化合物b2、b7的小鼠肾脏荷菌量实验结果(白念耐药菌感染)如图1所示,图1是化合物b2、b7的白念耐药菌感染小鼠肾脏荷菌量实验结果示意图。化合物b2、b7的小鼠生存期
实验结果(白念耐药菌感染)如图2所示,图2是化合物b2、b7的白念耐药菌感染小鼠生存期实验结果示意图。化合物b2、b7的小鼠肾脏荷菌量实验结果(耳念珠菌感染)如图3所示。图3是化合物b2、b7的耳念珠菌感染小鼠肾脏荷菌量实验结果示意图。
[0253]
体内实验结果表明,对于白念耐药菌感染动物模型,化合物b2、b7可显著降低小鼠的肾脏荷菌量(p《0.01),并且口服有效,其中化合物b7可有效延长感染小鼠的生存期(p《0.05)。对于耳念珠菌感染动物模型,化合物b2、b7经口服给药可显著降低小鼠肾脏荷菌量(p《0.05)。
[0254]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1