一种芬太尼类蛋白偶联物及其应用
1.本发明属于生物医药技术领域,具体涉及一种芬太尼类蛋白偶联物及其应用。
背景技术:
2.药物使用障碍(ouds)是一个全球性的健康问题,在过去的十年里,在北美和全世界,阿片类药物使用障碍的患病率和与阿片类药物相关的过量发生率以惊人的速度增长。在美国,至少有250万人患有ouds,并出现很多阿片类药物相关的致命过量事件。芬太尼是一种有效的合成阿片类药物,合法用作处方止痛药。然而,芬太尼表现出明显的滥用倾向,因为它通过激活大脑中的μ-阿片受体(mor)而产生愉悦的感觉,过度的mor激活会导致呼吸抑制,这可能是致命的。因此,当芬太尼从不受管制的来源摄入时,它会造成过量使用的重大风险。此外,芬太尼合成的简便性使得非法生产和制造特制的药物类似物成为可能。这些类似物的药理学尚未得到适当的表征,这使得它们特别危险,特别是当某些修饰,甚至是甲基添加,可以提高一定的效力。已批准的对抗药物过量的方法包括阿片受体激动剂美沙酮、部分激动剂丁丙诺啡、拮抗剂纳曲酮和纳洛酮等。这些药物安全有效,但由于副作用、管理障碍以及滥用和转移注意力的可能性,临床结果仍然不太理想,因此急切需要安全、持久和有效的治疗方法,以对抗由芬太尼引起的毒性和过量。
3.近年来,一种免疫疗法来对抗芬太尼及新精神活性物质的有害和成瘾作用。该策略的基础涉及蛋白质-药物结合物的主动疫苗接种,以产生体内免疫拮抗剂,从而有效地将作用部位的目标药物浓度降至最低。芬太尼特异性抗体减少热板的抗伤害感受和芬太尼诱导的呼吸抑制,这些研究涉及用多克隆抗体被动免疫或用弗氏完全佐剂皮内主动接种或其他佐剂腹膜内给药,这在人类中可能不可行。michael d.等人研究了在小鼠和大鼠体内肌肉注射的芬太尼疫苗的效力。以芬太尼为基础的半抗原与天然的钥孔血蓝蛋白(klh)载体蛋白或gmp grade亚单位klh(sklh)结合开发的蛋白偶联物,通过使用碳二亚胺化学的四甘氨酸接头分别产生 f-klh和f-sklh。与对照组相比,用f-klh免疫的小鼠和大鼠都具有较低的芬太尼诱导的抗伤害性。与对照组相比,静脉注射芬太尼后,用f-klh免疫的大鼠脑中芬太尼浓度较低。在另一组大鼠中,在一系列累积皮下芬太尼剂量下,f-sklh减少了芬太尼引起的热板的抗伤害性、呼吸抑制和心动过缓。
4.随着新类似物的出现及其供应,芬太尼类似物的使用迅速发生变化。但这类疫苗还处于新药开发时期,疫苗使用的剂量以及产生抗体的浓度还有待提高。因此,研究新型芬太尼疫苗成为对抗芬太尼使用障碍和毒性的可行策略。
技术实现要素:
5.本发明的目的是提供一种以4-苯基氨基-1-苯乙基哌啶作母核的芬太尼类蛋白偶联物及其应用。
6.为了实现上述目的,本发明采用以下技术方案:
7.一种芬太尼类半抗原,其结构式如式
ⅰ‑
a所示:
[0008][0009]
式中,r1为c1~c20的烷基、酰胺基;
[0010]
r2为c1~c20的烷基、酰胺基、烯键、任选地含有1-3个选自n、o或s的杂原子和c
1-6
‑ꢀ
烷基、c
3-8-杂环烷基的3-8元饱和或不饱和环;
[0011]
优选的,r1选自c1~c4的烷基,r2为c1~c4的烷基。
[0012]
一种芬太尼类抗原,其结构式如式ⅰ所示:
[0013][0014]
式中:r1选自c1~c20的烷基或酰胺基;
[0015]
r2选自c1~c20的烷基、酰胺基、烯键、任选地含有1-3个选自n、o或s的杂原子和c
1-6-烷基、c
3-8-杂环烷基的3-8元饱和或不饱和环,优选为c1~c20的烷基、酰胺基;
[0016]
protein为血蓝蛋白(klh)或霍乱毒素亚基b(ctb)。
[0017]
进一步地,所述芬太尼类抗原选自以下结构式中的一个:
[0018][0019]
上述芬太尼类抗原的制备方法,如下式所示:
[0020][0021]
其中:r1为c1~c20的烷基、酰胺基;r2为c1~c20的烷基、酰胺基;protein为血蓝蛋白(klh) 或霍乱毒素亚基b(ctb)。
[0022]
具体地:包括以下步骤:
[0023]
步骤1,化合物1与ch3ocor2cocl反应,得化合物2;
[0024]
步骤2,化合物2经水解反应得化合物3
[0025]
步骤3,化合物3与氨基化合物缩合分别得化合物4和5,4和5分别脱保护基得化合物
ⅰ‑
a。
[0026]
步骤4,化合物
ⅰ‑
a与蛋白偶联得式ⅰ所示的芬太尼类蛋白偶联物。
[0027]
作为本技术的优选技术方案,所述的步骤1为,0℃条件,将有机溶剂加入化合物1,再加入ch3ocor2cocl,移至室温反应,得到化合物2。其中有机溶剂为无水二氯甲烷。
[0028]
作为本技术的优选技术方案,化合物2经水解脱甲酯得式化合物2,其中,所用碱为2m 氢氧化锂或氢氧化钠水溶液。
[0029]
作为本技术的优选技术方案,化合物
ⅰ‑
a在4℃下经n-酰基琥珀酰亚胺活化,加入三乙胺,再与蛋白偶联,4℃透析24小时得式ⅰ类系列化合物。其中,蛋白为klh或ctb。
[0030]
上述芬太尼类半抗原或芬太尼类抗原在制备治疗阿片类药物的滥用及阿片类药物使用障碍的药物中的应用。
[0031]
本发明达到的有益效果:
[0032]
(1)本发明提供的一类母核为4-苯基氨基-1-苯乙基哌啶的芬太尼类半抗原,结构新颖,制备方法简单;
[0033]
(2)本发明引入霍乱霉素b亚基蛋白,将芬太尼类半抗原与该蛋白连接,使其能够更好的作用于t细胞,产生更多的抗体。
[0034]
(3)本发明采用蛋白质-药物结合物的主动疫苗接种,产生体内免疫拮抗剂,从而有效地将作用部位的目标药物浓度降至最低。最终,芬太尼疫苗可降低芬太尼的成瘾性和过量用药潜力。
附图说明
[0035]
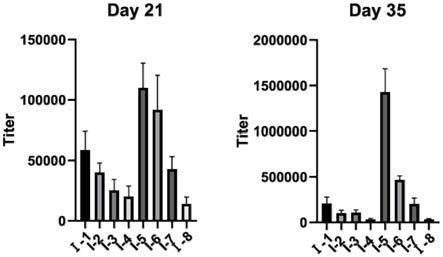
图1为elisa试验检测疫苗产生的抗体滴度值。
[0036]
图2为i-5(bsa)蛋白的表征-maldi-tof。
具体实施方式
[0037]
下面结合附图和具体实施例对本发明作进一步详细说明,但不应理解为对本发明的限制。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改或替换,均属于本发明的范围。实施例中未注明具体条件的实验方法及未说明配方的试剂均为按照本领域常规条件。
[0038]
在以下实施例中,采用bsa为蛋白,用于抗体滴度的检测。
[0039]
实施例1
[0040]
制备
ⅰ‑
1a
[0041]
1、合成化合物2(6-氧代-6-((1-苯乙基哌啶-4-基)(苯基)氨基)己酸甲酯)
[0042]
将4-苯基氨基-1-苯乙基哌啶(1.0g,3.56mmol)溶于无水二氯甲烷(5ml)中,将反应移至0℃,加入吡啶(950.43μl,31.60mmol)搅拌后,缓慢滴入甲基脂肪酰氯 (695.7μl,4.63mmol)搅拌10mⅰn后,移至室温反应2h,。反应完全后,用饱和nahco3 溶液淬灭,乙酸乙酯萃取,水层用etoac萃取3次。合并有机相,用饱和nacl溶液洗涤,无水na2so4干燥。硅胶柱层析纯化(石油醚/乙酸乙酯=4:1),得到淡黄色固体980mg,产率为86%。1h nmr(300mhz,chloroform-d)δ7.52
–
7.34(m,3h),7.28(dt,j=6.9,1.4hz,2h),7.20 (tt,j=7.9,1.5hz,3h),7.15
–
7.02(m,2h),4.71(tt,j=12.2,4.0hz,1h),3.66(s,3h),3.11
–ꢀ
2.98(m,2h),2.83
–
2.71(m,2h),2.63
–
2.48(m,2h),2.29
–
2.15(m,4h),1.96(t,j=7.0hz, 2h),1.90
–
1.77(m,2h),1.68
–
1.43(m,6h).
[0043]
2、合成化合物3(6-氧代-6-((1-苯乙基哌啶-4-基(苯基)氨基)己酸)
[0044]
将化合物2(200mg,0.489mmol)溶于1ml甲醇,全部溶解后,加入氢氧化锂水溶液(2ml) 室温搅拌2h后,用1n hcl溶液调ph至3~4,滤出白色固体190mg,产率为80%。1h nmr (300mhz,chloroform-d)δ7.47(d,j=6.7hz,3h),7.31(q,j=5.7,5.3hz,3h),7.26
–
7.21(m, 2h),7.18
–
7.03(m,2h),4.86
–
4.72(m,1h),3.61(d,j=11.9hz,2h),3.25
–
3.10(m,4h),2.91 (t,j=12.3hz,2h),2.25(t,j=6.9hz,2h),2.21
–
1.93(m,6h),1.55(qt,j=10.1,5.3hz,4h).
[0045]
3、合成化合物
ⅰ‑
1a
[0046]
步骤一:3-(6-氧代-6-((1-苯乙基哌啶-4-基)(苯基)氨基)六氨基)丙酸叔丁酯
(化合物5)
[0047]
将化合物3(100mg,0.244mol)溶于dcm(1ml)中,加入1-乙基-(3-二甲基氨基丙基) 碳酰二亚胺盐酸盐(edc)(70.39mg,0.367mmol),1-羟基苯并三唑(hobt) (49.61mg,0.367mmol),n,n-二异丙基乙胺(63.59μl,0.367mmol),搅拌30mⅰn后,加入β
‑ꢀ
丙氨酸叔丁酯盐酸盐(66.70mg,0.292mmol),反应完全后,加入水,乙酸乙酯萃取(3次),合并有机相,用饱和食盐水洗涤,无水na2so4干燥。硅胶柱层析纯化(石油醚/乙酸乙酯=1:1),得到白色固体69mg,产率为59%。1h nmr(300mhz,chloroform-d)δ7.46
–
7.31(m,3h), 7.31
–
7.21(m,2h),7.21
–
7.10(m,3h),7.06(dd,j=7.4,2.0hz,2h),6.28(t,j=6.0hz,1h), 4.67(tt,j=12.1,3.9hz,1h),3.44(q,j=6.1hz,2h),3.07
–
2.96(m,2h),2.79
–
2.60(m,2h), 2.60
–
2.48(m,2h),2.42(t,j=6.1hz,2h),2.24
–
2.01(m,4h),1.98
–
1.73(m,4h),1.62
–
1.48 (m,4h),1.44(s,9h).
[0048]
步骤二:3-(6-氧代-6-((1-苯乙基哌啶-4-基)(苯基)氨基)六酰胺基)丙酸(化合物
ⅰ‑
1a)
[0049]
将化合物
ⅰ‑
1-5(100mg,0.186mmol)溶于三氟乙酸和二氯甲烷的1:1混合溶液(500μl),反应2h,旋干溶剂得淡黄色油状液体97mg,产率为90%。1h nmr(300mhz,dmso-d6)δ7.87 (t,j=5.6hz,1h),7.53(q,j=7.8,7.0hz,3h),7.34(d,j=6.9hz,2h),7.30
–
7.23(m,5h),4.81
ꢀ–
4.68(m,1h),4.05(q,j=7.1hz,2h),3.20(p,j=5.5,4.6hz,6h),2.98(dd,j=11.2,5.7hz, 2h),2.36(t,j=6.9hz,2h),1.95(t,j=6.8hz,3h),1.85(t,j=6.9hz,2h),1.70
–
1.51(m,2h), 1.48
–
1.25(m,5h).
[0050]
将化合物
ⅰ‑
1a(20mg,0.037mmol)溶于dmf-h2o(10%)(500μl)中,加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc)(42.97mg,0.224mmol)、n-羟基琥珀酰亚胺(nhs) (8.59mg,0.0746mmol)、三乙胺(31.59μl,0.224mmol),室温反应6h后,旋干溶剂,加入 bsa-pbs溶液(6mg/ml,300μl),4℃搅拌12h,透析24h,将反应液冻干,得白色粉末。
[0051]
klh偶联疫苗和上述制备方法一致,最终得
ⅰ‑
1。
[0052]
实施例2-5
[0053]
参照实施例1的制备方法制备化合物
ⅰ‑
3a、
ⅰ‑
4a、
ⅰ‑
6a、
ⅰ‑
8a。
[0054]
[0055][0056]
实施例6
[0057]
制备化合物
ⅰ‑
2a:
[0058]
步骤一:4-(6-氧代-6-((1-苯乙基哌啶-4-基)(苯基)氨基)六酰胺基)丁酸甲酯(化合物4)
[0059]
将化合物3(200mg,0.489mmol)溶于dcm(1ml)中,加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc)(140.77mg,0.734mmol),1-羟基苯并三唑(hobt) (99.23mg,0.734mmol),n,n-二异丙基乙胺(170.55μl,0.734mmol),搅拌30min后,加入4
‑ꢀ
氨基丁酸甲酯盐酸盐(90.24mg,0.586mmol),反应完全后,加入水,乙酸乙酯萃取(3次),合并有机相,用饱和食盐水洗涤,无水na2so4干燥。硅胶柱层析纯化(石油醚/乙酸乙酯=1:1),得到白色固体160mg,产率为70%。1h nmr(300mhz,chloroform-d)δ7.48
–
7.38(m,3h), 7.37
–
7.15(m,6h),7.11(dt,j=7.4,1.7hz,2h),6.13(s,1h),4.71(tt,j=12.2,3.9hz,1h),3.71 (s,3h),3.32(q,j=6.6hz,2h),3.05(d,j=11.3hz,2h),2.77(dd,j=10.7,5.8hz,2h),2.58(dd, j=10.7,5.9hz,2h),2.41(t,j=7.3hz,2h),2.18(dt,j=13.4,8.5hz,4h),1.97(t,j=6.5hz, 2h),1.91
–
1.78(m,4h),1.68
–
1.41(m,6h).
[0060]
步骤二:4-(6-氧代-6-((1-苯乙基哌啶-4-基)(苯基)氨基)六氨基)丁酸(
ⅰ‑
2a)
[0061]
将步骤一的化合物(200mg,0.405mmol)溶于1ml甲醇,全部溶解后,加入氢氧化锂水溶液(2ml)室温搅拌2h后,用1n hcl溶液调ph至3~4,滤出白色固体170mg,产率为 84%。1h nmr(300mhz,dmso-d6)δ12.08(s,1h),7.81(t,j=5.6hz,1h),7.49(q,j=7.7,7.1 hz,3h),7.38
–
7.19(m,7h),4.72(t,j=12.4hz,1h),3.52(d,j=11.9hz,2h),3.23
–
3.06(m, 4h),2.99(p,j=6.0,5.2hz,4h),2.19(t,j=7.4hz,2h),2.00
–
1.87(m,4h),1.83(t,j=6.9hz, 2h),1.73
–
1.51(m,4h),1.36(dtd,j=19.5,11.9,9.3,5.2hz,4h).
[0062]
将化合物
ⅰ‑
2a(20mg,0.041mmol)溶于dmf-h2o(10%)(500μl)中,加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc)(46.60mg,0.246mmol)、n-羟基琥珀酰亚胺(nhs) (9.32mg,0.082mmol)、三乙胺(33.78μl,0.246mmol),室温反应6h后,旋干溶剂,加入bsa-pbs 蛋白溶液(6mg/ml,300μl,4℃搅拌12h,透析24h,将反应液冻干,得白色粉末。
[0063]
klh偶联半抗原和上述制备方法一致,最终得
ⅰ‑
2。
[0064]
实施例7-8
[0065]
参照实施例6的制备方法制备化合物
ⅰ‑
5a、
ⅰ‑
7a。
[0066][0067]
试验例
[0068]
elisa测试抗体滴度和竞争性ic
50
值
[0069]
在96孔板上,每孔涂1.5μg/ml fent-bsa包被蛋白,在4℃下过夜,并用pbst溶液洗涤3次。在室温下用脱脂牛奶封闭1小时后,5
×
pbs溶液洗涤,从1:1000开始,将接种的小鼠血清1%bsa溶液中以1:1的比例连续稀释,12个梯度加入到96孔板。在室温下孵育 1.5小时后,将板洗涤5次,然后加入在1%bsa的1:600稀释的驴抗小鼠igg辣根过氧化物酶(hrp),并在室温下孵育1.5小时。进行5
×
洗涤和3,3',5,5'-四甲基联苯胺(tmb)底物的添加(thermo pierce),然后在添加tmb 5分钟后添加2m h2so4。让板孵育10分钟,然后在 450nm处读取其吸光度od450。p/n值大于2的值定为抗体滴度值。
[0070]
竞争性elisa也以类似的方式进行,但增加了一个步骤:将ic
50
稀释度的血清与60um 至0.1nm的游离芬太尼稀释液(11个3倍稀释液)在fent-bsa包被板中孵育2小时。在 graphpad prism中,将吸光度值归一化为每个样品的最高吸光度值,归一化响应—可变斜率方程以确定中点滴度和标准误差。未接种疫苗的小鼠不含有任何可检测的抗芬太尼效价。
[0071]
实验结果:
[0072]
表一:化合物对芬太尼的竞争性ic50值
[0073][0074]
由表一结果可知,连续稀释的游离芬太尼与固定化药物的半抗原竞争抗体,来自 fent-klh(i)免疫小鼠的抗体对芬太尼有很高的亲和力,产生的结合曲线具有低纳摩尔ic
50
值。
[0075]
由图一结果可知,不同蛋白偶联物在接种21天和35天有显著的抗体滴度值,并随着时间的延长,滴度值在上升,说明蛋白偶联物具有一定的免疫疗效。
[0076]
由竞争性elisa试验表明,疫苗i-5对芬太尼有较高的亲和力,并在体内也有显著的抗体滴度值。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1