一种2-正戊基环戊-2-烯酮的制备方法与流程
1.本发明涉及香料生产技术领域,更具体地说,本发明涉及一种2-正戊基环戊-2-烯酮的制备方法。
背景技术:
2.2-正戊基环戊-2-烯酮,英文名:2-pentyl circularpenketone 2-olefin,为无色或淡黄色油状液体,主要用于合成香料二氢茉莉酮酸甲酯。
3.专利ep1577287中公开了以2-亚戊基环戊酮为原料,以盐酸、3-甲基吡啶为催化剂,在正丁醇的水溶液中发生异构反应,最后经烧碱中和后制备得到2-正戊基环戊-2-烯酮,产物收率为83%。该方法以大量的廉价盐酸为异构化试剂,但产物收率低,盐酸、正丁醇的单耗较高,设备腐蚀严重,部分盐酸与正丁醇发生取代反应生成氯代正丁烷,且反应完毕后,剩余的盐酸与烧碱反应生成难以处理废盐,存在严重的环保问题。
4.专利cn106946705中报道了以氢溴酸为催化剂,在正丁醇中回流反应2h,经5%碳酸钠水溶液中和后,精馏得到2-正戊基-2-环戊-2-烯酮,产物收率85%。该专利工艺较为简洁,但使用了过量的氢溴酸,设备腐蚀严重;同时大量的氢溴酸被正丁醇消耗转化为溴丁烷,致使氢溴酸的利用率不高;而且产物收率不高,有较多的脚料产生。
5.专利ep2269971中提出了一种以钯碳为催化剂催化双键异构的方法。该方法以正癸烷为溶剂,在氢气氛围中活化催化剂后,于190℃下反应6h,产物2-正戊基环戊-2-烯酮的气相色谱选择性为96%。该方法使用了价格昂贵的贵金属催化剂钯碳,且反应温度过高,易使热稳定性较差的原料和产物发生聚合反应,生成高沸点的杂质,降低了产物收率,而且反应条件苛刻,难以实现工业化。
技术实现要素:
6.为了克服现有技术的上述缺陷,本发明提供了一种工艺简洁、反应条件温和、收率高、废盐少、易产业化的2-正戊基环戊-2-烯醇的合成工艺。
7.为实现上述目的,本发明提供如下技术方案:一种2-正戊基环戊-2-烯酮的制备方法,包括以下步骤:
8.步骤一:将2-亚戊基环戊酮、溶剂、助剂、质子酸加入反应容器中,搅拌,加热至一定温度下,保温一定时间,保温至反应完毕;
9.步骤二:降温至室温后,加入一定量碱中和,再经水洗、脱溶、减压蒸馏后得到成品2-戊基环戊-2-烯酮。
10.在上述方案中,采用一锅法将全部物料一起加入反应容器中,其加入顺序对反应结果没有明显影响,一锅法的投料方法使助剂、质子酸在反应体系中的浓度可以维持在较高水平,有利于提高反应速率、缩短反应时间。
11.在一种优选的实施方式中,所述步骤一中质子酸为盐酸、氢溴酸、硫酸、磷酸、硝酸、亚磷酸、焦磷酸、苯膦酸、二苯基磷酸、磷酸二氢钠、磷酸二氢钾、磷酸二氢铵、氯化铵、溴
化铵、硫酸氢铵、硝酸铵或草酸铵中的一种,所述质子酸的用量为2-亚戊基环戊酮质量的0.05%~20%,优选的,质子酸为硫酸、磷酸、硝酸、氢溴酸或溴化铵中的一种,所述质子酸的用量为2-亚戊基环戊酮质量的0.1%~5%。
12.在上述方案中,质子酸在异构反应中起到关键作用,原料2-亚戊基环戊酮中的双键与质子酸中的氢离子发生加成反应,生成碳正离子,从而引发双键的异构反应。当无质子酸存在时,在相当长的反应时间内,只有微量的异构产物2-正戊基环戊-2-烯酮产生。本发明减少了质子酸的用量,降低了质子酸的单耗,采用催化量的质子酸即可使反应进行完全。催化量的质子酸在反应体系中的浓度非常小,可减缓质子酸对金属材质的腐蚀。同时,减少了中和质子酸产生的废盐量,使该工艺更加安全环保。
13.在一种优选的实施方式中,所述步骤一中助剂为环丁砜、乙二醇二甲醚、乙二醇二乙醚、乙二醇二丁醚、二乙二醇二甲醚、二乙二醇二乙醚、二乙二醇丙醚、二乙二醇己醚、三乙二醇甲醚、三乙二醇乙醚、三乙二醇丁醚、四乙二醇二甲醚、四乙二醇乙醚、五乙二醇甲醚或六乙二醇甲醚中的一种,所述助剂的用量为2-亚戊基环戊酮质量的0.05%~20%,优选的,助剂为二乙二醇二丁醚、环丁砜、三乙二醇甲醚、三乙二醇乙醚、三乙二醇丁醚或四乙二醇二甲醚中的一种,所述助剂的用量为2-亚戊基环戊酮质量的0.1%~5%。
14.在上述方案中,助剂在异构反应中可发挥两方面的作用,一方面,助剂将质子酸从水中转移至油相中,使质子酸在油相中与原料2-亚戊基环戊酮进行异构反应;另一方面,因助剂与原料2-亚戊基环戊酮双键质子化形成的碳正离子产生范德华作用力,生成的氢键可以稳定该碳正离子,减少其发生水合、聚合等副反应,有利于提高产物2-正戊基环戊-2-烯酮的选择性。当无助剂存在且只有催化量质子酸时,在相当长的反应时间内,只有少量的异构产物2-正戊基环戊-2-烯酮产生。本发明采用助剂的用量为催化量,大大降低了原辅料的成本。
15.在一种优选的实施方式中,所述步骤一中溶剂为甲苯、二甲苯、三甲苯、四甲苯、氯苯、邻二氯苯、异丙苯、二苯甲烷、正己烷、环己烷、正庚烷、正辛烷、正壬烷、正癸烷、乙酸乙酯、乙酸丙酯、乙酸丁酯、乙酸戊酯、四氢呋喃或二氧六环中的一种,所述溶剂的用量为2-亚戊基环戊酮质量的0.5~10倍,优选的,溶剂为氯苯、二甲苯、三甲苯、四甲苯或正庚烷中的一种,所述溶剂的用量为2-亚戊基环戊酮质量的2~7倍。
16.在上述方案中,添加溶剂的目的是将原料2-亚戊基环戊酮稀释,原料的浓度过高会发生酸引发的羟醛缩合、双键聚合等副反应。加入溶剂后原料2-亚戊基环戊酮的浓度降低,可大大减少上述副反应,提高产物2-正戊基环戊-2-烯酮的收率。
17.在一种优选的实施方式中,所述步骤一中反应温度为80~160℃,保温时间为0.5~15h,优选的,反应温度为110~130℃,保温时间为3~9h。
18.在上述方案中,双键异构反应在室温时无法进行,需要在一定温度下才可以进行反应。其主要副反应为原料2-亚戊基环戊酮和产物2-正戊基环戊-2-烯酮的羟醛缩合、双键的聚合;若反应温度过高,会加剧上述副反应,使反应收率降低。另外,不同溶剂对反应温度有直接相关的影响,比如极性较弱的溶剂在较高反应温度下的收率较高,而极性较强的溶剂在较低反应温度下的收率会较高。反应时间与反应温度相关,在较高反应温度下的反应时间较短,在较低反应温度下的反应时间较长。
19.在一种优选的实施方式中,所述步骤二中碱中和时加入的碱为氢氧化钠、氢氧化
钾、氢氧化锂、氢氧化铯、氢氧化镁、氢氧化钡、碳酸钠、碳酸钾、碳酸锂、碳酸氢钠或碳酸氢钾中的一种,所述碱中和后的ph为5~9,优选的,碱中和时加入的碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠或碳酸氢钾中的一种,碱中和后的ph为6~8。
20.在上述方案中,因异构产物2-正戊基环戊-2-烯酮在酸性条件下的热稳定性较差,会发生质子酸引发的羟醛缩合、双键聚合等副反应,所以需要将反应液用碱中和至中性,使异构产物在较为稳定的中性环境中进行进一步的后处理。
21.在一种优选的实施方式中,所述步骤二中水洗是向中和液中加入适量的水,搅拌均匀后静置分层,上层为油相,下层为水相;所述步骤二中脱溶是指将水洗后的上层油相加入脱溶瓶中,在负压氛围中进行蒸馏,回收溶剂,所述步骤二中减压蒸馏是指回收完溶剂后,更换蒸馏接收瓶,在负压氛围中继续进行蒸馏,接收异构产物2-正戊基环戊-2-烯酮成品。
22.在上述方案中,在异构反应完毕后,经过简单的中和、水洗、脱溶、减压蒸馏即可得到异构产物成品2-正戊基环戊-2-烯酮。助剂留存于减压蒸馏后的釜液中,该釜液可以不经纯化直接套用于下一批反应。因异构反应存在羟醛缩合、双键聚合等副反应,产生的高沸杂质也会留存于减压蒸馏后的釜液中。随着釜液套用次数的增加,釜液量不断增加,一般情况下,釜液套用的次数为10~15次,但套用第15次时反应选择性仍未明显降低,水洗的目的是将中和产生的无机盐溶于水中,通过分液分出下层含盐水相,达到除盐的目的。
23.本发明的技术效果和优点:
24.1、本发明使用催化剂量的助剂和质子酸,工艺条件温和,大大减轻了设备腐蚀,提高设备的安全性。采用催化剂量的酸,显著地减少了含盐废水生成量,降低了三废,绿色环保,有利于二氢茉莉酮酸甲酯的工业化大生产;
25.2、本发明使用“一锅法”的反应方式,工艺流程简洁、操作简便,异构产物2-正戊基环戊-2-烯酮的收率高。
具体实施方式
26.下面将结合本发明中的实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
27.实施例1
28.将200g 2-亚戊基环戊酮、100g甲苯、0.1g盐酸、0.1g二乙二醇二丁醚加入反应容器中,搅拌,加热至80℃,保温15h至反应完毕。降温至室温后,加入0.8g 5%氢氧化钠溶液中和,中和液ph=5。向中和液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂甲苯97g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮182.75g,含量94.49%,收率88.10%,釜液剩余17.35g。
29.实施例2
30.将200g 2-亚戊基环戊酮、100g二甲苯、0.2g氢溴酸、0.2g二乙二醇二丁醚加入反应容器中,搅拌,加热至110℃,保温9h至反应完毕。降温至室温后,加入1.5g 5%氢氧化钾
溶液中和,中和液ph=6。向中和液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂二甲苯98g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮189.36g,含量94.79%,收率91.58%,釜液剩余10.84g。
31.实施例3
32.将100g 2-亚戊基环戊酮、400g二甲苯、1g溴化铵、1g二乙二醇二丁醚加入反应容器中,搅拌,加热至120℃,保温6h至反应完毕。降温至室温后,向反应液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂二甲苯398g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮95.80g,含量94.97%,收率92.84%,釜液剩余5.20g。
33.本实验为最佳方案,针对本实验进行了釜液的套用,即将实施例3的蒸馏釜液替代助剂二乙二醇二丁醚投入反应中进行异构反应。由套用数据可知,随着蒸馏釜液的套用批次增加,蒸馏釜液的质量也越来越大,套用至15批次时的蒸馏釜液已经达到88.54g,但产品的收率并未明显降低,产品收率依然维持在较高水平。考虑到蒸馏釜液质量增加过大,未继续套用蒸馏釜液。蒸馏釜液套用数据如表1。
34.表1催化剂套用数据
35.序号t/℃t/h釜液/g产品/g含量/%收率/%套用1120611.0095.2095.3992.66套用2120616.1695.8595.3693.27套用3120621.0596.1195.3593.51套用4120625.1996.8694.0392.94套用5120631.0795.1194.9692.17套用6120636.4695.6194.8492.53套用7120642.3395.1395.0792.29套用8120647.2896.0594.0892.20套用9120653.9594.3295.9092.30套用10120660.0994.8695.8192.74套用11120665.9295.1795.6892.92套用12120671.3795.5594.9292.55套用13120677.3295.0495.5592.67套用14120682.3395.9994.5392.60套用15120688.5494.8095.6992.56
36.对比实施例1无质子酸参与的异构反应
37.将100g 2-亚戊基环戊酮、400g二甲苯、1g二乙二醇二丁醚加入反应容器中,搅拌,加热至120℃,保温6h至反应完毕。降温至室温后,向反应液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂二甲苯398g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮0.78g,含量98.87%,收率0.79%,釜液剩余100.22g。
38.对比实施例2无助剂参与的异构反应
39.将100g 2-亚戊基环戊酮、400g二甲苯、1g溴化铵加入反应容器中,搅拌,加热至120℃,保温6h至反应完毕。降温至室温后,向反应液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂二甲苯397g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮2.35g,含量98.69%,收率2.37%,釜液剩余97.65g。
40.由上述两个对比实验可知,在无质子酸参与的异构反应中,产物收率只有0.79%,仍有大量未反应的原料2-亚戊基环戊酮残留在蒸馏釜液中。在无助剂参与的异构反应中,产物收率为2.37%,略高于无质子酸参与的异构反应,因此质子酸比助剂更加关键。
41.实施例4
42.将100g 2-亚戊基环戊酮、200g三甲苯、0.5g硫酸、1g环丁砜加入反应容器中,搅拌,加热至120℃,保温6h至反应完毕。降温至室温后,加入21.2g 5%碳酸钠溶液中和,中和液ph=7。向反应液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂三甲苯198g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮94.95g,含量94.33%,收率91.39%,釜液剩余6.05g。
43.实施例5
44.将100g 2-亚戊基环戊酮、400g四甲苯、5g磷酸、5g环丁砜加入反应容器中,搅拌,加热至130℃,保温3h至反应完毕。降温至室温后,加入197g 5%碳酸钾溶液中和,中和液ph=8,分出下层水相。向上层油相中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂四甲苯392g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮93.61g,含量95.54%,收率91.25%,釜液剩余11.39g。
45.实施例6
46.将100g 2-亚戊基环戊酮、700g氯苯、2g硝酸、0.5g乙二醇二乙醚加入反应容器中,搅拌,加热至110℃,保温9h至反应完毕。降温至室温后,加入25g 5%氢氧化钠溶液中和,中和液ph=7。向反应液中加入50g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂氯苯686g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮92.08g,含量95.89%,收率90.10%,釜液剩余8.42g。
47.实施例7
48.将200g 2-亚戊基环戊酮、400g邻二氯苯、5g亚磷酸、0.1g乙二醇二甲醚加入反应容器中,搅拌,加热至80℃,保温15h至反应完毕。降温至室温后,加入64.4g 5%氢氧化镁溶液中和,中和液ph=6,分出下层水相。向上层油相中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂邻二氯苯392g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮184.25g,含量95.53%,收率89.80%,釜液剩余15.85g。
49.实施例8
50.将200g 2-亚戊基环戊酮、400g异丙苯、1g焦磷酸、0.2g二乙二醇己醚加入反应容器中,搅拌,加热至110℃,保温9h至反应完毕。降温至室温后,加入19.1g 5%氢氧化钡溶液
中和,中和液ph=7。向反应液中加入50g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂异丙苯393g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮180.52g,含量95.71%,收率88.15%,釜液剩余19.68g。
51.实施例9
52.将100g 2-亚戊基环戊酮、400g异丙苯、1g苯磷酸、1g三乙二醇甲醚加入反应容器中,搅拌,加热至120℃,保温6h至反应完毕。降温至室温后,加入3.04g 5%氢氧化锂溶液中和,中和液ph=8。向反应液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂异丙苯395g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮94.63g,含量94.21%,收率90.97%,釜液剩余6.37g。
53.实施例10
54.将200g 2-亚戊基环戊酮、200g正己烷、5g乙二醇二丁醚、20g二苯基磷酸加入反应容器中,搅拌,加热至80℃,保温14h至反应完毕。降温至室温后,加入272g 5%氢氧化铯溶液中和,中和液ph=6,分出下层水相。向上层油相中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在常压下进行脱溶,回收溶剂和轻组分201g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮181.73g,含量95.40%,收率88.45%,釜液剩余23.27g。
55.实施例11
56.将100g 2-亚戊基环戊酮、400g环己烷、20g磷酸二氢钠、1g二乙二醇二甲醚加入反应容器中,搅拌,加热至80℃,保温0.5h至反应完毕。降温至室温后,加入244g 5%碳酸锂溶液中和,中和液ph=7,分出下层水相。向上层油相中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在常压下进行脱溶,回收溶剂环己烷390g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮91.43g,含量94.40%,收率88.07%,釜液剩余9.57g。
57.实施例12
58.将50g 2-亚戊基环戊酮、500g正庚烷、20g磷酸二氢钾、5g三乙二醇乙醚加入反应容器中,搅拌,加热至100℃,保温8h至反应完毕。降温至室温后,向反应液中加入40g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂正庚烷490g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮47.39g,含量94.14%,收率91.05%,釜液剩余7.61g。
59.实施例13
60.将100g 2-亚戊基环戊酮、400g正辛烷、5g磷酸二氢铵、20g二乙二醇丙醚加入反应容器中,搅拌,加热至110℃,保温9h至反应完毕。降温至室温后,向反应液中加入50g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂正辛烷392g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮90.78g,含量95.53%,收率88.49%,釜液剩余29.22g。
61.实施例14
62.将100g 2-亚戊基环戊酮、400g正壬烷、1g氯化铵、1g三乙二醇丁醚加入反应容器
中,搅拌,加热至120℃,保温6h至反应完毕。降温至室温后。向反应液中加入100g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂正壬烷390g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮91.82g,含量94.90%,收率88.91%,釜液剩余9.18g。
63.实施例15
64.将100g 2-亚戊基环戊酮、400g正癸烷、5g溴化铵、5g四乙二醇二甲醚加入反应容器中,搅拌,加热至130℃,保温3h至反应完毕。降温至室温后,向反应液中加入100g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂正癸烷392g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮91.80g,含量95.37%,收率89.34%,釜液剩余13.20g。
65.实施例16
66.将100g 2-亚戊基环戊酮、400g乙酸乙酯、1g硫酸氢铵、1g四乙二醇二甲醚加入反应容器中,搅拌,加热至80℃,保温15h至反应完毕。降温至室温后,向反应液中加入100g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂乙酸乙酯392g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮92.23g,含量95.15%,收率89.56%,釜液剩余8.76g。
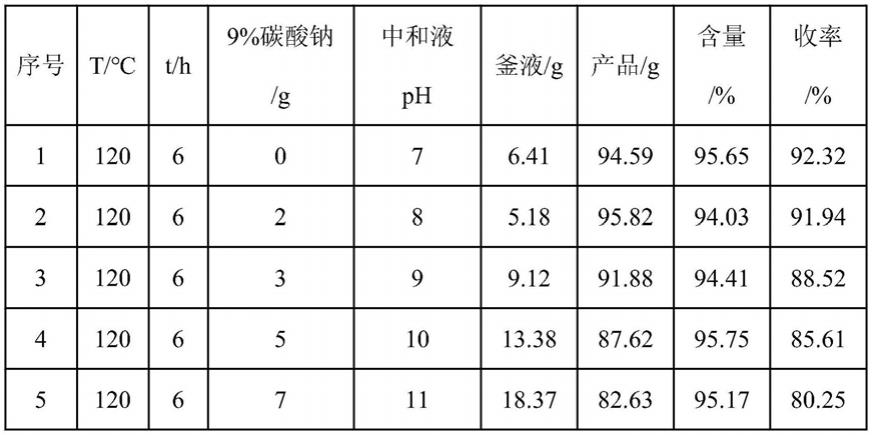
67.实施例17
68.将100g 2-亚戊基环戊酮、400g乙酸丙酯、5g硝酸铵、1g四乙二醇丁醚加入反应容器中,搅拌,加热至90℃,保温12h至反应完毕。降温至室温后,向反应液中加入30g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂乙酸丙酯392g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮90.95g,含量94.84%,收率88.01%,釜液剩余14.05g。
69.实施例18
70.将100g 2-亚戊基环戊酮、400g乙酸丁酯、1g草酸铵、1g五乙二醇甲醚加入反应容器中,搅拌,加热至110℃,保温9h至反应完毕。降温至室温后,向反应液中加入100g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂乙酸丁酯395g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮90.94g,含量95.62%,收率88.74%,釜液剩余10.05g。
71.实施例19
72.将100g 2-亚戊基环戊酮、400g乙酸戊酯、5g盐酸、5g六乙二醇甲醚加入反应容器中,搅拌,加热至150℃,保温1h至反应完毕。降温至室温后,加102g 5%碳酸氢钾溶液中和,中和液ph=7。向反应液中加入50g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂乙酸戊酯390g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮92.02g,含量95.49%,收率89.67%,釜液剩余12.98g。
73.实施例20
74.将100g 2-亚戊基环戊酮、400g二氧六环、5g氯化铵、5g六乙二醇甲醚加入反应容器中,搅拌,加热至100℃,保温10h至反应完毕。降温至室温后,向反应液中加入40g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空
下进行脱溶,回收溶剂二氧六环392g。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮92.10g,含量95.74%,收率89.98%,釜液剩余12.90g。
75.实施例21碱中和后不同ph下的异构反应
76.将100g 2-亚戊基环戊酮、400g二甲苯、1g溴化铵、1g二乙二醇二丁醚加入反应容器中,搅拌,加热至120℃,保温6h至反应完毕。降温至室温后,加入一定量9%碳酸钠溶液中和。向中和液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂二甲苯。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮,成品含量、收率、釜液质量如表2。
77.表2碱中和后不同ph下的异构反应数据
[0078][0079]
由表2数据可知,不加9%碳酸钠溶液中和的中和液的ph=7,即在室温下该反应液为中性。随着9%碳酸钠溶液的加入,中和液的ph被提高,水洗后不能将过量的碱清除彻底,产品在碱性条件下发生聚合副反应,导致成品收率逐渐降低。
[0080]
实施例22不同温度下的异构反应
[0081]
将100g 2-亚戊基环戊酮、400g二甲苯、1g溴化铵、1g二乙二醇二丁醚加入反应容器中,搅拌,加热至一定温度,保温至反应完毕。降温至室温后,向反应液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂二甲苯。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮,成品含量、收率、釜液质量如表3。
[0082]
表3不同温度下的异构反应
[0083]
[0084][0085]
在反应温度为80℃时,反应速率显著下降,保温15h后仍有较多原料未反应完全,产品的分离收率只有58.63%。随着反应温度的继续升高,产品收率先升高后下降。当温度在110-130℃范围内时,产品收率最高。
[0086]
实施例23不同量质子酸下的异构反应
[0087]
将100g 2-亚戊基环戊酮、400g二甲苯、1g二乙二醇二丁醚和一定质量的溴化铵加入反应容器中,搅拌,加热至120℃,保温至反应完毕。降温至室温后,向反应液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂二甲苯。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮,成品含量、收率、釜液质量如表4。
[0088]
表4不同量质子酸下的异构反应
[0089][0090][0091]
由表4可知,当溴化铵的质量为0.1g时,反应速率大大降低,需要延长反应时间至
12h才能反应完毕。因反应时间延长,副反应增多,导致产品收率较低。随着溴化铵用量的增加,产品收率先升高后下降。溴化铵用量较高时,酸性增加,反应速率较快,副反应亦增多,致使产品收率降低。当溴化铵的质量为1-2g时,产品收率最高。
[0092]
实施例24不同量助剂下的异构反应
[0093]
将100g 2-亚戊基环戊酮、400g二甲苯、1g溴化铵和一定质量的助剂加入反应容器中,搅拌,加热至120℃,保温至反应完毕。降温至室温后,向反应液中加入25g自来水进行水洗,静置分层,分出上层油相。将上层油相转移至脱溶瓶中,在循环水真空泵抽真空下进行脱溶,回收溶剂二甲苯。脱溶完毕后,在油泵抽真空下进行减压蒸馏,收集馏分得成品2-戊基环戊-2-烯酮,成品含量、收率、釜液质量如表5。
[0094]
表5不同量助剂下的异构反应
[0095][0096]
由表5可知,由于助剂二乙二醇二丁醚的量较少时(0.1g),反应速率显著降低,需要延长反应时间至14h才能反应完毕。因反应时间延长,副反应增多,导致产品收率略微下降。随着二乙二醇二丁醚用量的增加,产品收率趋于稳定。
[0097]
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1