一种卟啉-富勒烯分子开关化合物的制备方法
1.本发明属于功能有机超分子设计与合成技术领域,具体是涉及一种卟啉-富勒烯分子开关化合物的制备方法。
背景技术:
2.具有三维球状结构的富勒烯分子,自发现至今吸引了科学家的广泛关注。由于其独特的空间结构,因而具有许多优异的性质,如三维刚性电子离域、低重组能、低还原势和长电荷分离态等。富勒烯是优异的电子受体材料,基于富勒烯的pc
61
bm和pc
71
bm的有机太阳能电池受体材料已实现商用。卟啉环是高度共轭的体系,可以作为一种优良的电子给体。卟啉与富勒烯之间的给-受体相互作用,导致卟啉与富勒烯之间可以相互吸引,从而发生光致电子转移等光物理过程,对于人工光合作用的模拟研究具有重要意义。
3.间苯二酚杯[4]芳烃分子开关化合物在一定外界刺激,如温度、ph和金属离子的作用下,会发生构象的转变。发生构象转变时,不同分子手臂的距离则会发生变化。当间苯二酚杯[4]芳烃分子开关化合物同时桥联起卟啉和富勒烯分子时,卟啉与富勒烯之间的空间距离可受外界刺激发生变化。
[0004]
目前对卟啉与富勒烯之间的光致电子转移、能量转移及光物理过程的研究主要聚焦在较短共价键连接上,对于卟啉与富勒烯之间通过非共价键形成的能量转移、电子转移过程了解不足。
技术实现要素:
[0005]
为了克服现有阶段中存在的不足,本发明的目的之一在于提供一种卟啉-富勒烯分子开关化合物,它既可以用于研究卟啉与富勒烯之间非共价键形成的能量转移、电子转移过程,又可用于研究长共价键之间的光物理过程,形成一种新型的功能有机超分子体系。
[0006]
为实现该目的,本发明采用了以下技术方案:
[0007]
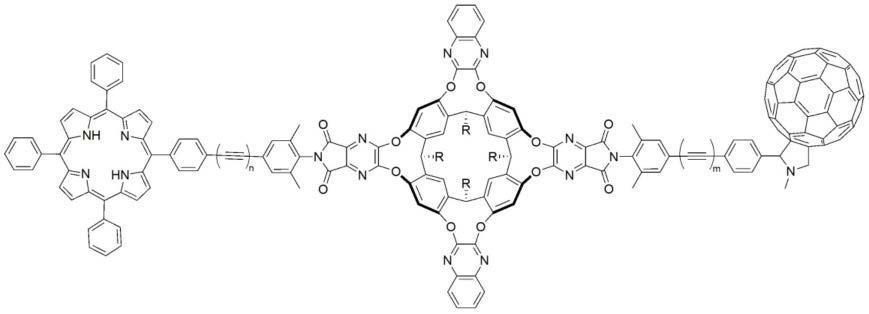
一种卟啉-富勒烯分子开关化合物,其分子结构通式如下所示:
[0008][0009]
通式中r为各个独立的c1-c11疏水性长链基团;
[0010]
通式中x为乙炔基或频哪醇硼酸酯基;
[0011]
通式中y为乙炔基或频哪醇硼酸酯基;
[0012]
通式中m为0或1;
[0013]
通式中n为0或1。
[0014]
上式中,卟啉和富勒烯皆通过刚性链与间苯二酚杯[4]芳烃孔穴化合物相连,从而形成一个可受外界刺激调控、具有开关性质的孔穴化合物。
[0015]
本发明的另一目的在于提供一种卟啉-富勒烯分子开关化合物的制备方法,为实现该目的,本发明采用了以下技术方案:
[0016]
卟啉-富勒烯分子开关化合物的制备方法,制备路线如图1所示,依次包括如下步骤:
[0017]
步骤
①
:化合物1和化合物2通过suzuki偶联或者sonogashira偶联反应得到一边是氨基一边是醛基的化合物3。化合物1与化合物2的摩尔比为0.8~1∶0.8~1,反应温度为室温,反应时间为36~48h。
[0018]
步骤
②
:化合物3和化合物4在吡啶与草酰氯条件下发生反应得到醛基手臂化合物5。化合物3与化合物4的摩尔比为1∶1.2~1.5,反应温度首先为室温,其次加热到60℃,再冷却至50℃。反应时间为24~36h。
[0019]
步骤
③
:化合物6和化合物7通过suzuki偶联或者sonogashira偶联反应得到末端是氨基的四苯基卟啉化合物8。化合物6与化合物7的摩尔比为1∶1.5~3。反应温度为95℃,反应时间为12~24h。
[0020]
步骤
④
:化合物8和化合物4在吡啶与草酰氯条件下发生反应得到卟啉手臂化合物9。化合物8与化合物4的摩尔比为1∶1.2~1.5,反应温度首先为室温,其次加热到80℃,再冷却至70℃。反应时间为12~18h。
[0021]
步骤
⑤
:化合物5和化合物10在碳酸铯条件下发生亲核取代反应得到带一个醛基手臂的化合物11。化合物5与化合物10的摩尔比为1∶1~1.5。反应温度为60℃,反应时间为10~14h。
[0022]
步骤
⑥
:化合物11和c
60
及肌氨酸发生prato反应得到带一个c
60
的孔穴化合物12。化合物11与c
60
及肌氨酸的摩尔比为1∶1.5~3∶8~12。反应温度为110℃,反应时间为10~14h。
[0023]
步骤
⑦
:化合物12和化合物9在碳酸铯条件下发生亲核取代反应得到卟啉-富勒烯分子开关化合物13。化合物12与化合物9的摩尔比为1∶1.1~1.3。反应温度为60℃,反应时间为10~14h。
[0024]
作为本发明的卟啉-富勒烯分子开关化合物的制备方法的进一步改进:
[0025]
所述步骤
①
的具体操作为:将化合物1和化合物2按照0.8~1∶0.8~1的摩尔比溶解在无水四氢呋喃中,通入氩气20~40min,迅速加入催化剂,继续通氩气10~30min,将反应瓶密封,注射三乙胺,室温搅拌36~48h,利用薄层色谱根据,待原料点基本消失,抽滤,减压除去四氢呋喃和三乙胺,再将混合物过柱分离,得到化合物3。
[0026]
所述步骤
②
的具体操作为:将化合物3和化合物4按照1∶1.2~1.5的摩尔比溶解在无水四氢呋喃中,60℃反应10~14h,利用薄层色谱跟踪化合物3反应完毕时,将反应混合物冷却至室温,将无水吡啶注射进反应体系,草酰氯滴加进反应体系中,待草酰氯滴加完毕时,升温至50℃,反应12~24h。利用薄层色谱根据,待产生的新点基本不再变化时,停止反
应。抽滤,减压除去四氢呋喃,再将混合物过柱分离,得到化合物5。
[0027]
所述步骤
③
的具体操作为:将化合物6和化合物7按照1∶1.5~3的摩尔比溶解在无水dmf中,通入氩气20~40min,迅速加入催化剂,继续通氩气10~30min,将反应瓶密封,95℃反应12~24h,用薄层色谱根据,待原料点基本消失,减压除去dmf,再将混合物过柱分离,得到化合物8。
[0028]
所述步骤
④
的具体操作为:将化合物8和化合物4按照1∶1.2~1.5的摩尔比溶解在无水1,4-二氧六环中,80℃反应10~14h,利用薄层色谱跟踪化合物8反应完毕时,将反应混合物冷却至室温,将无水吡啶注射进反应体系,草酰氯滴加进反应体系中,待草酰氯滴加完毕时,升温至70℃,反应4~8h。利用薄层色谱根据,待产生的新点基本不再变化时,停止反应。抽滤,减压除去1,4-二氧六环,再将混合物过柱分离,得到化合物9。
[0029]
所述步骤
⑤
的具体操作为:将化合物化合物5和化合物10按照1∶1~1.5的摩尔比溶解在无水四氢呋喃中,升温至60℃,待温度稳定后加入碳酸铯,反应时间为10~14h。用薄层色谱根据,待原料点基本反应完毕时,停止反应。抽滤,减压除去四氢呋喃,再将混合物过柱分离,得到化合物11。
[0030]
所述步骤
⑥
的具体操作为:将化合物11和c
60
及肌氨酸按照1∶1.5~3∶8~12的摩尔比溶解在甲苯中,升温至110℃,反应时间为10~14h。用薄层色谱根据,待原料点基本反应完毕时,停止反应。抽滤,减压除去甲苯,再将混合物过柱分离,得到化合物12。
[0031]
所述步骤
⑦
的具体操作为:将化合物12和化合物9按照1∶1.1~1.3的摩尔比溶解在无水四氢呋喃中,升温至60℃,待温度稳定后加入碳酸铯,反应时间为10~14h。用薄层色谱根据,待原料点基本反应完毕时,停止反应。抽滤,减压除去四氢呋喃,再将混合物过柱分离,得到化合物13。
[0032]
本发明提出的一种卟啉-富勒烯分子开关化合物,其结构通式中卟啉和富勒烯通过刚性链与间苯二酚杯[4]芳烃相连。与现有技术相比,本发明的有益效果表现在:
[0033]
1)本发明利用间苯二酚杯[4]芳烃将卟啉和富勒烯桥联起来,形成具有开关性质的分子,实现了同一分子内卟啉与富勒烯之间的距离可调,拓展了分子内光物理过程变化的研究种类。
[0034]
2)本发明制备的卟啉-富勒烯分子开关化合物可通过构象转变应用于超分子络合作用的探针。
[0035]
3)本发明的卟啉-富勒烯分子开关化合物可应用在光动力治疗(pdt)、非线性光学等领域。
附图说明
[0036]
图1为卟啉-富勒烯分子开关化合物的制备路线图。
[0037]
图2为实施例制备的卟啉-富勒烯分子开关化合物的1h nmr图。
[0038]
图3为实施例制备的卟啉-富勒烯分子开关化合物的
13
c nmr图。
[0039]
图4为实施例制备的卟啉-富勒烯分子开关化合物的cosy nmr图。
[0040]
图5为实施例制备的卟啉-富勒烯分子开关化合物的noesy nmr图。
[0041]
图6为实施例制备的卟啉-富勒烯分子开关化合物的hmbc nmr图。
[0042]
图7为实施例制备的卟啉-富勒烯分子开关化合物的高分辨质谱图。
[0043]
图8为实施例制备的卟啉-富勒烯分子开关化合物在氘代三氟乙酸以及氘氧化钠重水溶液调控下的1h nmr图。
[0044]
图9为实施例制备的卟啉-富勒烯分子开关化合物在三氟乙酸调控下的荧光发射光谱图。
具体实施方式
[0045]
本实施例以一种卟啉-富勒烯分子开关化合物13-1为例,结合制备路线图(附图1所示)介绍其结构以及具体的制备方法。
[0046]
步骤
①
[0047]
干燥的100ml三颈烧瓶中,将化合物1-1(18.29mmol)和化合物2(18.44mmol)溶解在无水四氢呋喃(100ml)中,通入氩气30min,迅速加入双三苯基膦二氯化钯(0.1425mmol)和碘化亚铜(0.5251mmol),继续通氩气30min,将反应瓶密封,注射三乙胺(28.70mmol),室温搅拌48h,利用薄层色谱根据,待原料点基本消失,抽滤,减压除去四氢呋喃和三乙胺,以石油醚∶二氯甲烷=1∶1(v∶v)将混合物过柱分离,得到化合物3(52.85%)。
[0048]
所述化合物1-1为所述化合物1中的x是乙炔基。
[0049]
所述化合物1-1的结构式如下所示:
[0050][0051]
步骤
②
[0052]
干燥的250ml三颈烧瓶中,将化合物3(2.856mmol)和化合物4(3.726mmol)溶解在无水四氢呋喃(80ml)中,60℃反应12h,利用薄层色谱跟踪化合物3反应完毕时,将反应混合物冷却至室温,将无水吡啶(9.469mmol)注射进反应体系,草酰氯(4.317mmol)滴加进反应体系中,待草酰氯滴加完毕时,升温至50℃,反应18h。利用薄层色谱根据,待产生的新点基本不再变化时,停止反应。抽滤,减压除去四氢呋喃,以石油醚∶二氯甲烷=1∶1(v∶v)将混合物过柱分离,得到化合物5-1(81%)。
[0053]
所述化合物5-1为所述化合物5中的m=1。
[0054]
所述化合物5-1的结构式如下所示:
[0055][0056]
步骤
③
[0057]
干燥的100ml三颈烧瓶中,将化合物6-1(0.675mmol)和化合物7(1.5mmol)溶解在无水dmf(60ml)中,通入氩气30min,迅速加入四三苯基膦钯(0.108mmol),继续通氩气30min,将反应瓶密封,95℃搅拌12h,利用薄层色谱根据,待原料点基本消失,将反应液倒入水(1000ml)中,抽滤,
[0058]
干燥,以石油醚∶二氯甲烷=1∶1(v∶v)将混合物过柱分离,得到化合物8-1
(53.75%)。
[0059]
所述化合物6-1为所述化合物6中的y=频哪醇硼酸酯基。
[0060]
所述化合物6-1的结构式如下所示:
[0061][0062]
所述化合物8-1为所述化合物8中的n=0。
[0063]
所述化合物8-1的结构式如下所示:
[0064][0065]
步骤
④
[0066]
干燥的100ml三颈烧瓶中,将化合物8-1(0.354mmol)和化合物4(0.457mmol)溶解在无水1,4-二氧六环(60ml)中,90℃反应12h,利用薄层色谱跟踪化合物8-1反应完毕时,将反应混合物冷却至室温,将无水吡啶(0.708mmol)注射进反应体系,草酰氯(1.168mmol)滴加进反应体系中,待草酰氯滴加完毕时,升温至80℃,反应6h。利用薄层色谱根据,待产生的新点基本不再变化时,停止反应。将反应液倒入水(500ml)中,抽滤,干燥,以石油醚∶二氯甲烷=1∶1(v∶v)将混合物过柱分离,得到化合物9-1(81%)。
[0067]
所述化合物9-1为所述化合物9中的n=1。
[0068]
所述化合物9-1的结构式如下所示:
[0069][0070]
步骤
⑤
[0071]
干燥的50ml反应管中,将化合物5-1(0.1333mmol)和化合物10-1(0.1671mmol)溶解在无水四氢呋喃中,升温至60℃,待温度稳定后加入碳酸铯(0.138mmol),反应时间为12h。用薄层色谱根据,待原料点基本反应完毕时,停止反应。抽滤,减压除去四氢呋喃,以二氯甲烷∶乙酸乙酯=100∶3(v∶v)将混合物过柱分离,得到化合物11-1(62%)。
[0072]
所述化合物10-1为所述化合物10中的r=—ch2ch2ch2ch2ch2ch3。
[0073]
所述化合物11-1为所述化合物11中的m=1,r是正己基(即—c6h
13
)。
[0074]
所述化合物11-1的结构式如下所示:
[0075][0076]
步骤
⑥
[0077]
干燥的100ml三颈烧瓶中,将化合物11-1(0.1mmol)和c
60
(0.2mmol)及肌氨酸(0.3mmol)溶解在甲苯(50ml)中,升温至110℃,反应时间为12h。用薄层色谱根据,待原料点基本反应完毕时,停止反应。抽滤,减压除去甲苯,以二氯甲烷∶乙酸乙酯=100∶1(v∶v)将混合物过柱分离,得到化合物12-1(61%)。
[0078]
所述化合物12-1为所述化合物12中的m=1,r是正己基(即—c6h
13
)。
[0079]
所述化合物12-1的结构式如下所示:
[0080][0081]
步骤
⑦
[0082]
干燥的50ml三颈烧瓶中,将化合物12-1(0.1mmol)和化合物9-1(0.13mmol)溶解在无水四氢呋喃(40ml)中,升温至60℃,待温度稳定后加入碳酸铯(0.1mmol),反应时间为12h。用薄层色谱根据,待原料点基本反应完毕时,停止反应。抽滤,减压除去四氢呋喃,以石油醚∶二氯甲烷=1∶4(v∶v)将混合物过柱分离,得到化合物13-1(53%)。
[0083]
所述化合物13-1为所述化合物13中的m=1,n=0,r是正己基(即—c6h
13
)。
[0084]
所述化合物13-1的结构式如下所示:
[0085][0086]
上述实施例制得的目标化合物13-1的1h nmr、
13
c nmr和高分辨质谱分别如附图2、3和4所示。
[0087]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,随着目标化合物13(结构通式如下)的m、n、r的不同选择,将会制备出若干不同结构的卟啉-富勒烯分子开关化合物。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
[0088][0089]
示例1(卟啉-富勒烯分子开关化合物13-2):
[0090]
通式中,m=1,n=1,r是正己基。
[0091]
示例2(卟啉-富勒烯分子开关化合物13-3):
[0092]
通式中,m=0,n=0,r是正己基。
[0093]
示例3(卟啉-富勒烯分子开关化合物13-4):
[0094]
通式中,m=0,n=1,r是正己基。
[0095]
上述不同的卟啉-富勒烯分子开关化合物(13-2至13-4),其制备方法及反应机理与化合物13-1较为相似,在此不再一一赘述。
[0096]
综上所述,本发明提供了一种卟啉-富勒烯分子开关化合物的制备方法。该功能分子在外界刺激下具有开关作用,有望应用于人工光合作用光反应阶段的模拟、光动力治疗(pdt)以及非线性光学等领域。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1