一类基于硫代五甲川菁染料的无重原子三重态光敏剂、其制备方法和应用
1.本发明涉及一种高效三重态光敏剂,特别涉及一类基于硫代五甲川菁染料的无重原子三重态光敏剂、其制备方法和应用。
背景技术:
2.三重态光敏剂是指被光激发后能够通过系间窜越产生三重态的一类化合物。具有长寿命三重态激发态的光敏剂更有利于电子转移或能量转移来充分利用三重态性质,可广泛应用于光动力治疗(pdt)、光催化、三重态-三重态湮灭(tta)上转换、和光伏发电等,在能源和生命科学领域有良好的应用前景。为了实现高效的系间窜越,利用分子设计策略来构建能够有效产生三重态的化合物成为该领域的研究热点。目前,利用重原子效应仍然是最方便、最有效的方法,但生物毒性大和三重态寿命短等缺点极大限制了三重态能量的进一步应用于其他领域。相反,无重原子三重态光敏剂可以在不受重原子效应影响下获得长三重态寿命。然而,分子结构与系间窜越性质之间的关系还没有完全明确的规律,系统地设计高效的无重原子三重态光敏剂仍然是一个挑战。
3.目前,一些不依赖于重原子且可以有效促进系间窜越的策略已经被开发出来,并且已成功应用于萘酰亚胺、芘、氟硼吡咯等染料中,但是一般难以克服荧光量子产率低、合成路线复杂、吸收波长处于可见区、三重态寿命短、结构高度疏水等缺点。菁染料具有合成简洁、近红外吸收可调、荧光量子产率高、生物相容性好等优点,由于不具备三重态性质起初被广泛应用于荧光探针中。为了设计适用于光动力治疗的三重态光敏剂,选择本身性质优异的菁染料进行合理修饰是一条新的途径。目前对于菁染料的分子设计均选择在共轭链中位进行修饰,这大大限制了光敏剂的发展,迫切需要一系列新的分子来指导无重原子三重态光敏剂的设计。
4.光动力治疗作为一种新兴的癌症治疗手段被广泛研究,其主要原理是使无毒的光敏剂进入目标靶点,随后利用与之匹配的光照激发光敏剂产生具有细胞毒性的活性氧物质来破坏病变组织,从而达到治疗的目的。然而,目前使用的光敏剂仍然存在许多缺陷,理想的光敏剂至少需要具备以下性质:1.在近红外区拥有优异的光子捕获能力;2.高的活性氧量子产率和长的三重激发态寿命;3.良好的生物相容性、光稳定性和适宜的水溶性;4重要的亚细胞器(线粒体、内质网等)定位性和肿瘤靶向性。至今,具有以上所有优点的三重态光敏剂尚未开发问世。
技术实现要素:
5.针对菁染料在三重态光敏剂方面技术的不足,本发明公开了一类基于硫代五甲川菁染料的无重原子三重态光敏剂,该类光敏剂在生命科学领域,尤其是光动力治疗方面具有良好的应用前景。首先根据菁染料确定的中位基团和苯环上的功能基团确定荧光发射和近红外吸收波长,进一步通过在硫代五甲川菁染料上连接不同杂环进行修饰有效限制了非
辐射跃迁,在尽可能不改变荧光发射和近红外吸收波长的情况下,增强了系间窜越并延长了三重态寿命。此类分子具有良好的水溶性、线粒体定位性和光稳定性,能够在近红外光激发下产生活性氧,进行癌细胞或活体肿瘤的光动力治疗。
6.本发明第一方面提供了一类基于硫代五甲川菁染料的无重原子三重态光敏剂,其如结构通式i所示:
[0007][0008]
通式中:
[0009]
x为:cl、br、i中的一种;
[0010]
r1、r2各自独立的选自h、甲基、甲氧基、f、cl、氰基、氨基、羧基、硝基、磺酸基中的一种;
[0011]
r3、r4各自独立的选自-c
mh2m-r6中的一种,其中m选自1-5的整数,包括1、2、3、4、5,更优选地,m=1-3;
[0012]
r6选自呋喃基、噻吩基、异噁唑基、异噻唑基、吡唑基、噁唑基、噻唑基、咪唑基、哒嗪基、嘧啶基、吡嗪基、苯并呋喃基、苯并噻唑基、吲哚基、喹啉基、异喹啉基、嘌呤基、噻吩[3,2-b]并噻吩基、带有单个或多个二级取代基的上述基团衍生结构中的一种;
[0013]
r5选自醛基、苯基、萘基、蒽基、带有单个或多个二级取代基的上述基团衍生结构中的一种;
[0014]
本发明第二方面在于保护一类基于硫代五甲川菁染料的无重原子三重态光敏剂的制备方法,包括如下合成步骤:
[0015]
步骤s1:2-甲基苯并噻唑衍生物与卤代烷基衍生物溶解于溶剂i中,升温至60-100℃过夜反应,得到固体即为2-甲基苯并噻唑季铵盐;
[0016]
步骤s2:0-5℃下,将三氯氧磷滴加到dmf中,搅拌1-4小时,随后加入溴乙酸,加热至80℃-100℃反应完全后,进行相应的后处理获得固体1即为r5取代基的丙二醛;其中,后处理分为两种情况:当s2加入时,向反应液中加入饱和naclo4溶液直至产生沉淀获得固体,随后将固体溶解于naoh并用盐酸将溶液调至酸性ph=1-3可得固体1;当s2加入溴乙酸时,向反应液中加入乙醇和高氯酸直至产生沉淀获得固体2,随后将固体2溶解于甲醇并用naoh调ph=11-14得固体3,将固体3溶于二氯甲烷中并用盐酸调ph=1-3,
最后利用二氯甲烷萃取并旋干获得固体1;
[0017]
步骤s3:将相应的2-甲基苯并噻唑衍生物季铵盐和带有r5取代基的丙二醛混合于溶剂ii,添加催化剂后升温至50-100℃反应完全后,通过柱层析纯化,除去溶剂后获得硫代五甲川菁染料。
[0018]
进一步优选的情况下,所述步骤s1中的2-甲基苯并噻唑衍生物与卤代烷基衍生物摩尔比为1:1-10;更为优选的是1-4。
[0019]
进一步优选的情况下,所述步骤s1中的溶剂i为乙腈、乙醇、甲苯、二甲苯中的至少一种。
[0020]
进一步优选的情况下,所述步骤s2中的三氯氧磷和乙酸衍生物的摩尔比为2-8:1。
[0021]
进一步优选的情况下,所述步骤s3中的2-甲基苯并噻唑衍生物季铵盐与带有r5取代基的丙二醛摩尔比为1-4:1。
[0022]
进一步优选的情况下,所述步骤s3中的溶剂为乙醇、甲醇、乙酸酐、乙酸中的至少一种。
[0023]
进一步优选的情况下,所述步骤s3中的催化剂为吡啶、哌啶、醋酸钠、碳酸钾中的至少一种;步骤s3中的催化剂与带有r5取代基的丙二醛摩尔比为3-20:1。
[0024]
进一步优选的情况下,所述步骤s3中柱层析纯化的洗脱剂配比为甲醇与二氯甲烷按体积比1:5-100。
[0025]
本发明第三方面在于保护一类基于硫代五甲川菁染料的无重原子三重态光敏剂的应用,主要涉及生命科学领域。
[0026]
进一步优选的情况下,所述应用包括在生物成像、近红外发光、联合治疗或光动力治疗等方面中的应用。
[0027]
这类基于硫代五甲川菁染料的无重原子三重态光敏剂具有优异的水溶性,能够被细胞快速摄取并定位于亚细胞器(普遍为线粒体)中,在近红外光激发下产生活性氧物质,并进一步诱导细胞凋亡。良好的生物相容性和优异的三重态性质展现了生命科学领域方面优异的应用前景。
[0028]
与现有技术相比,本发明具有下列有益效果:
[0029]
目前,基于菁染料的三重态光敏剂开发仍然普遍采用重原子效应,难以获得具有优异生物应用前景的三重态性质。少数基于菁染料的无重原子三重态光敏剂的分子设计通常在于共轭链中位进行修饰,这容易显著改变分子的光谱性质,尤其是由于荧光发射与三重态产生是竞争关系,因此通常难以同时保证。本发明首先选择优异的中位基团来确定良好的近红外吸收和荧光发射,并进一步通过在硫代五甲川菁染料侧链进行合理修饰有效改善了其三重态性质,从而在光动力治疗中充分发挥菁染料本身摩尔消光系数大、合成简洁、荧光发射强、生物相容性好等优点。更为重要的是,修饰前后,三重态性质有了质的改变,系间窜越从无到有,并且具有优异的系间窜越效率,单线态氧量子产率最高能够接近90%;三重激发态寿命对于普通三重态光敏剂提升2-3个数量级,即使对比其他无重原子三重态光敏剂也有4-5倍的提升。该类分子由于优异的水溶性,能够不依赖于纳米粒子直接进行细胞或活体的光动力治疗,并且获得了优异的细胞杀伤和肿瘤抑制效果,肿瘤抑制率达到85%。
附图说明
[0030]
图1为本发明实施例tcy5-s、tcy5-o和对比例tcy5-et在二氯甲烷中的紫外吸收光谱;
[0031]
图2为本发明实施例tcy5-s、tcy5-o和对比例tcy5-et在二氯甲烷中的荧光发射光谱;
[0032]
图3为本发明实施例tcy5-s、tcy5-o和对比例tcy5-et在水中的单线态氧捕获剂abda的紫外吸收衰减曲线;
[0033]
图4为本发明实施例tcy5-s、tcy5-o和对比例tcy5-et的三线态寿命测试图;
[0034]
图5为本发明实施例tcy5-s在mcf-7细胞中的线粒体定位图;
[0035]
图6为本发明实施例tcy5-s对mcf-7细胞在常氧或乏氧条件下的细胞毒性测试数据图;
[0036]
图7为不同实验组在活体肿瘤光动力治疗实验中的肿瘤体积变化曲线;
[0037]
图8为不同实验组在活体肿瘤光动力治疗实验中的小鼠体重变化曲线。
具体实施方式
[0038]
结合附图,下面对本发明的具体实施方式进行详细地描述,但应当理解本发明的保护范围并不受具体实施方式的限制。
[0039]
除非另有说明,本发明使用的原料均可通过市售获得,或者可通过本领域技术人员公知的方法简单制备得到。
[0040]
实施例1
[0041]
本实施例为光敏剂tcy5-s的制备方法,包括如下步骤:
[0042]
步骤1:2-甲基苯并噻唑季铵盐的合成
[0043][0044]
将2-甲基苯并噻唑(10mmol)与2-溴甲基噻吩(10mmol)溶解于乙腈(15ml)中,随后升温至80℃搅拌过夜。反应完全后,冷却至0℃并用乙醚洗涤,可得纯度足够的固体产物(7.5mmol,产率75%)。
[0045]
步骤2:合成中间体丁三醛
[0046][0047]
将溴乙酸(60mmol)加入到dmf(30ml)中,随后在冰浴下缓慢滴加pocl3(200mmol)并搅拌3h。将冰浴换成油浴后,继续在90℃下搅拌过夜。反应完成后,在冰浴下加入乙醇(25ml)、水(3ml)和高氯酸(16ml),溶液析出固体。将固体混合于甲醇(48ml)中,搅拌下缓慢加入naoh颗粒,溶液先变为澄清再变为浑浊,收集固体溶解于二氯甲烷中,并用盐酸调ph至1。最后,利用二氯甲烷萃取并旋干溶剂所得固体即为丁三醛(40mmol,产率67%)。
[0048]
步骤3:硫代五甲川菁染料的合成
[0049][0050]
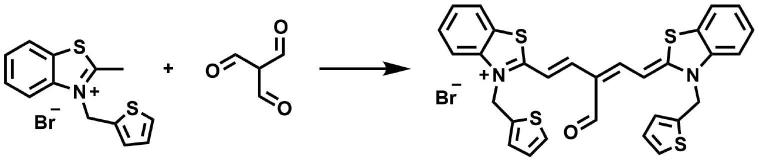
将2-甲基苯并噻唑季铵盐(0.5mmol)和丁三醛(0.25mmol)溶解于乙醇(15ml)中,随后加入一滴吡啶并升温至65℃反应。利用薄层色谱法检测反应进程,直至产物不再增加时,减压除去溶剂,并用柱层析(meoh:dcm=1:5-100)纯化可得带金属光泽的产物即为侧链为噻吩、中位为醛基的硫代五甲川菁染料tcy5-s。
[0051]
实施例2
[0052]
本实施例为光敏剂tcy5-o的制备方法,包括如下步骤:
[0053]
步骤1:2-甲基苯并噻唑季铵盐的合成
[0054][0055]
将2-甲基苯并噻唑(10mmol)与2-溴甲基呋喃(10mmol)溶解于乙腈(15ml)中,随后升温至70℃搅拌过夜。反应完全后,冷却至0℃并用乙醚洗涤,可得纯度足够的固体产物(9mmol,产率90%)。
[0056]
步骤2:合成中间体丁三醛
[0057][0058]
将溴乙酸(60mmol)加入到dmf(30ml)中,随后在冰浴下缓慢滴加pocl3(200mmol)并搅拌3h。将冰浴换成油浴后,继续在90℃下搅拌过夜。反应完成后,在冰浴下加入乙醇(25ml)、水(3ml)和高氯酸(16ml),溶液析出固体。将固体混合于甲醇(48ml)中,搅拌下缓慢加入naoh颗粒,溶液先变为澄清再变为浑浊,收集固体溶解于二氯甲烷中,并用盐酸调ph至3。最后,利用二氯甲烷萃取并旋干溶剂所得固体即为丁三醛(40mmol,产率67%)。
[0059]
步骤3:硫代五甲川菁染料的合成
[0060][0061]
将2-甲基苯并噻唑季铵盐(0.5mmol)和丁三醛(0.25mmol)溶解于乙醇(15ml)中,随后加入一滴吡啶并升温至65℃反应。利用薄层色谱法检测反应进程,直至产物不再增加时,减压除去溶剂,并用柱层析(meoh:dcm=1:5-100)纯化可得带金属光泽的产物即为侧链为呋喃、中位为醛基的硫代五甲川菁染料tcy5-o。
[0062]
对比例1
[0063]
本实施例为光敏剂tcy5-et的制备方法,包括如下步骤:
[0064]
步骤1:2-甲基苯并噻唑季铵盐的合成
[0065][0066]
将2-甲基苯并噻唑(10mmol)与碘乙烷(30mmol)溶解于乙腈(15ml)中,随后升温至80℃搅拌过夜。反应完全后,冷却至0℃并用乙醚洗涤,可得纯度足够的固体产物(5mmol,产率50%)。
[0067]
步骤2:合成中间体丁三醛
[0068][0069]
将溴乙酸(60mmol)加入到dmf(30ml)中,随后在冰浴下缓慢滴加pocl3(200mmol)并搅拌3h。将冰浴换成油浴后,继续在90℃下搅拌过夜。反应完成后,在冰浴下加入乙醇(25ml)、水(3ml)和高氯酸(16ml),溶液析出固体。将固体混合于甲醇(48ml)中,搅拌下缓慢加入naoh颗粒,溶液先变为澄清再变为浑浊,收集固体溶解于二氯甲烷中,并用盐酸调ph至2。最后,利用二氯甲烷萃取并旋干溶剂所得固体即为丁三醛(40mmol,产率67%)。
[0070]
步骤3:硫代五甲川菁染料的合成
[0071][0072]
将2-甲基苯并噻唑季铵盐(0.5mmol)和丁三醛(0.25mmol)溶解于乙醇(15ml)中,随后加入一滴吡啶并升温至65℃反应。利用薄层色谱法检测反应进程,直至产物不再增加时,减压除去溶剂,并用柱层析(meoh:dcm=1:5-100)纯化可得带金属光泽的产物即为侧链为乙基、中位为醛基的硫代五甲川菁染料tcy5-et。
[0073]
效果例1
[0074]
光敏剂tcy5-s、tcy5-o、tcy5-et的光谱测试:
[0075]
将tcy5-s、tcy5-o、tcy5-et分别溶解于二氯甲烷中,分别测试相应的紫外吸收和荧光发射谱图。
[0076]
如紫外吸收谱图(图1)所示,tcy5-s、tcy5-o、tcy5-et具有相似覆盖500-700nm的紫外吸收带,最大吸收波长为620nm,与600-800nm的“治疗窗口”重叠,表明侧链连接杂环不会影响其最大吸收波长。
[0077]
效果例2
[0078]
光敏剂tcy5-s、tcy5-o、tcy5-et的单线态氧产生能力测试:
[0079]
将光敏剂tcy5-s加入到含有3ml水溶液的石英皿中,并加入一定量的abda使其380nm处的吸光度约等于1。随后,利用近红外光(630nm,10mw/cm2)照射测试石英皿,每隔1分钟测试一次紫外吸收。tcy5-o和tcy5-et的单线态氧产生能力测试操作同tcy5-s,测试结
果整理如图3所示。
[0080]
根据图3,按照相关计算公式可得tcy5-s、tcy5-o、tcy5-et在水溶液中的单线态氧量子产率分别为50%,28%,2%。相比于对比例1的tcy5-et,tcy5-s和tcy5-o的单线态氧量子产率有一个明显的提升(分别提高25倍和14倍),说明在菁染料的侧链进行非共轭连接杂环可以有效促进化合物的系间窜越。
[0081]
效果例3
[0082]
光敏剂tcy5-s、tcy5-o、tcy5-et的三线态寿命测试:
[0083]
用574nm激光脉冲(1hz,100mj/脉冲,fwhm≈7ns)在室温下激发脱氧dcm中的tcy5-s、tcy5-o、tcy5-et,在620nm处测量了瞬态物质的衰减轨迹。
[0084]
如图4所示,tcy5-s具有超长的三线态寿命,高达472.4μs,这即使在无重原子三重态光敏剂中都是十分难得的,有利于克服肿瘤乏氧微环境,从而充分发挥tcy5-s的光动力治疗潜力。并且,tcy5-s和tcy5-o的三线态寿命相比于不进行侧链杂环修饰的tcy5-et均有一个倍数级别的提升,说明了该系列分子的有效性。
[0085]
效果例4
[0086]
光敏剂tcy5-s的亚细胞器定位实验:
[0087]
当mcf-7细胞密度在共聚焦培养皿中达到80%左右时,利用共聚焦激光扫描显微镜(clsm)进行线粒体共定位实验。首先用pbs清洗培养皿3次,然后加入2ml新鲜培养基、2μl tcy5-s(1mm)和线粒体绿色荧光探针(mito-tracker green)。培养皿置于培养箱中孵育30分钟,倒掉培养基,用pbs清洗3次后添加新鲜培养基。最后,使用clsm进行细胞荧光成像。
[0088]
如图5所示,tcy5-s的红色荧光与mito-tracker green的绿色荧光很好的重叠,皮尔森相关系数达到0.965,表示tcy5-s进入细胞后可以很好的定位于线粒体。线粒体是细胞重要的能量工厂,定位于线粒体有利于后续的光动力治疗。
[0089]
效果例5
[0090]
光敏剂tcy5-s在常氧或乏氧条件下对mcf-7的细胞毒性实验:
[0091]
将mcf-7细胞接种于96孔板中,置于培养箱中培养1-2天。当细胞密度达到90%左右时,在孔中加入100μl含不同浓度tcy5-s的新鲜培养基,浓度分别为2.5、1.25、0.63、0.31、0.16、0.08、0.04、0.02和0μm。在常氧或乏氧条件下,将细胞孵育2小时后光照(630nm,20mw/cm2)10分钟,再孵育12小时。上述治疗后,将原先培养基更换为含有mtt(0.5mg/ml)的新鲜培养基并放置在培养箱中进一步培养4小时。最后,向每孔中加入100μl dmso溶解甲臜晶体。最后,用酶标仪测定各孔在570nm(od)和630nm(odk)下的吸光度,并按照相关公式计算细胞存活率。每次向孔中加入新溶液前,用棉花将之前的溶液从孔中除去。
[0092]
如图6所示,tcy5-s在常氧和乏氧条件下均能对细胞造成杀伤,并且ic
50
低至0.25μm(常氧)和0.47μm(乏氧),这表明高的单线态氧量子产率和长的三重激发态寿命使tcy5-s具有克服缺氧限制的肿瘤光动力治疗能力。
[0093]
效果例6
[0094]
光敏剂tcy5-s的活体肿瘤光动力治疗实验:
[0095]
预先扩增4t1细胞,选择雌性小鼠腋窝部位皮下注射含5
×
106个4t1细胞的100μl pbs建立4t1肿瘤模型。肿瘤体积增大到200mm3后,将4t1荷瘤小鼠随机分为4组,每组3只。按组给予相应处理:(1)pbs-暗组:小鼠原位注射100μl pbs;(2)pbs-光组:小鼠原位注射100μ
l pbs,1小时后用近红外光源(630nm,50mw/cm2)照射15分钟;(3)tcy5-s-暗组:小鼠原位注射100μl含tcy5-s(50μm)的pbs;(4)tcy5-s-光组:小鼠原位注射100μl含tcy5-s(50μm)的pbs,1小时后用近红外光源(630nm,50mw/cm2)照射15分钟。每隔2天测量肿瘤的长度、宽度和小鼠的体重,根据以下公式计算:肿瘤体积=(宽度
×
宽度
×
长度)/2。
[0096]
如图7所示,仅注射了tcy5-s且光照的小鼠肿瘤体积维持在可控范围,而其他组别小鼠的肿瘤均生长至初始体积的12倍以上。这证明了tcy5-s能在近红外光激发下产生单线态氧,从而有效抑制乏氧肿瘤生长。
[0097]
如图8所示,活体肿瘤光动力治疗实验过程中,各组别小鼠体重均无异常变化,说明tcy5-s良好的生物相容性。
[0098]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,本领域的技术人员在本发明披露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求书的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1