一种人免疫组库测序的方法及试剂盒与流程
1.本技术涉及人免疫组库测序技术领域,特别是涉及一种人免疫组库测序的方法及试剂盒。
背景技术:
2.人免疫组库(immunerepertoire),指在任意指定时间点,个体循环系统中所有特异性不同的t淋巴细胞和b淋巴细胞克隆的总和。t细胞识别抗原并被激活的过程包括:抗原被抗原呈递细胞(如巨噬细胞)吞噬、加工、处理成多肽片段,以抗原肽-mhc复合物的形式表达于细胞表面,再被t细胞表面的t细胞受体(tcr)识别,从而激活机体免疫反应。免疫组库越丰富,机体越能有效抵抗细菌、病毒等病原体侵袭;反之,则越容易感染疾病。tcr是由两条不同肽链构成的异二聚体,大多数tcr,约95%左右,都是由α和β两条链组成,剩余的少数由γ和δ两条链组成,约5%左右。α和β两条链的v区(vα、vβ)又各有三个高变区cdr1、cdr2、cdr3,其中以cdr3变异最大,直接决定了tcr的抗原结合特异性。tcr的cdr3由v、d、j三个基因编码,在淋巴细胞的成熟过程中,通过v、d、j基因的重排形成了各种重组序列片段,这就解释了人类基因组及蛋白组学揭示的有限基因数目编码几乎无限蛋白种类的原因。tcr cdr3是t细胞识别抗原的主要部位。狭义上讲,tcr cdr3序列分析即可代表t细胞组库研究。
3.通常人免疫组库测序(ir-seq),指以t/b淋巴细胞为研究目标,以5’race技术或多重pcr扩增决定b细胞受体(bcr)或t细胞受体(tcr)多样性的互补决定区(cdr区),再结合高通量测序技术,全面评估免疫系统的多样性,深入挖掘免疫组库与疾病的关系。
4.然而,人免疫组库测序技术本身仍然具有一些缺陷。5’race技术是基于rna的扩增技术,其技术原理为,tcr/bcr的恒定区的通用引物序列向可变区扩增,然后引入的接头序列进行第二次无偏好的pcr扩增,其缺点在于:(1)使用rna样本作为扩增模板,rna有比dna更苛刻的提取条件,对实验环境和技术人员均有较高要求;(2)rna有易降解的缺陷,对原始组织样本的运输和保存均有严格的条件;(3)在实验操作上更加繁琐复杂。
5.多重pcr扩增虽然使用dna为作为原始模板,通过设计根据tcr v、d、j区域设计特异性引物,可以直接用于基因组dna,但是其缺陷也比较明显:(1)多重pcr引物设计难度高,扩增产物难以覆盖全部亚型;(2)现有技术操作过程复杂,多重pcr扩增结束、纯化后,需要进行末端修复,加a碱基及末端修饰,进行建库,操作比较繁琐;(3)多重pcr扩增会涉及到未发生重排或者正在发生重排的v、d、j区域片段,造成pcr扩增产物范围覆盖几十到上千bp长度片段,在此步需要对片段进行分选,保留较小有效片段,通常片段分选使用凝胶电泳切胶回收的方式操作,需花费较多的实验时间,同时对有效片段造成一定损失;操作上也比较繁琐;(4)tcrβ链cdr3的扩增不管以dna还是rna为模板,多重pcr特异性扩增阶段都无法避免出现偏好性扩增,较多的扩增循环数还会积累大量的扩增错误,不利于多样性的分析,易造成错误判读。
6.因此,如何减少或校正pcr扩增错误或人为因素导致的假阳性问题,仍然是人免疫组库测序的研究重点。
技术实现要素:
7.本技术的目的是提供一种改进的人免疫组库测序的方法及试剂盒。
8.为了实现上述目的,本技术采用了以下技术方案:
9.本技术的一方面公开了一种人免疫组库测序的方法,包括以下步骤:
10.配制反应体系,采用第一引物对模板核酸进行一次延伸,得到互补链;第一引物由5’端到3’端依序包括测序平台上游引物结合区、唯一性标识符和靶标特异性上游引物序列;并且,第一引物中,测序平台上游引物结合区和靶标特异性上游引物序列中的碱基t替换为脱氧尿嘧啶,测序平台上游引物结合区对应测序平台的上游测序引物的3’末端;
11.第一引物延伸结束后,向反应体系中加入第二引物,利用第二引物对第一引物延伸的互补链进行一次延伸,获得由测序平台上游引物结合区、唯一性标识符、靶标序列和测序平台下游引物结合区组成的产物;第二引物由5’端到3’端依序包括测序平台下游引物结合区和靶标特异性下游引物序列,测序平台下游引物结合区对应测序平台的下游测序引物的3’末端;
12.第二引物延伸结束后,向反应体系中加入udg/ung酶消化脱氧尿嘧啶,从而消化第一引物以及第一引物的延伸链;
13.udg/ung酶消化完成后,向反应体系中加入第三引物,利用第三引物和第二引物对第二引物延伸的产物进行pcr扩增富集,获得模板核酸的所有扩增子都添加相同唯一性标识符的产物;第三引物为第一引物的测序平台上游引物结合区自5’端起的全部或部分序列,并且,第三引物中的碱基t不被替换为脱氧尿嘧啶;
14.对pcr扩增富集获得的所有扩增子都添加相同唯一性标识符的产物进行测序文库构建和测序,即完成人免疫组库测序;
15.其中,靶标特异性上游引物序列和靶标特异性下游引物序列为针对人t细胞受体编码基因设计的全覆盖其cdr3区域编码基因序列的特异性引物序列。
16.本技术的方法中,“进行一次延伸”是指利用引物退火杂交到靶标序列上后,只进行这条引物的延伸即可,不进行再次的变性、退火。这样能够确保一个模板核酸母链被一个唯一的umi标记。当然,在第二引物加入后,虽然设计的是第二引物进行退火杂交、延伸;但是,这个时候,第一引物也会退火杂交、延伸;不过,由于第二引物的退火杂交、延伸也是只“进行一次延伸”;这种情况下,由测序平台上游引物结合区、唯一性标识符、靶标序列和测序平台下游引物结合区组成的产物同样只有最开始第一引物延伸标记umi的母链。最后,在第三引物和第二引物的指数倍pcr扩增富集下,也只有最开始第一引物延伸标记的umi母链的扩增子能够被指数倍富集。并且,在第三引物和第二引物进行pcr扩增富集之前,利用udg/ung酶消化去除了第一引物,也避免了第一引物在新的一轮pcr扩增中再次引入新的umi。本技术的模板核酸,可以是dna或cdna。
17.需要说明的是,本技术的方法,通过对第一引物、第二引物和第三引物进行特殊设计,依序向反应体系中添加三种引物,可以实现对某一个模板核酸母链的所有扩增子链都添加相同的umi,这对于突变检测尤其重要。例如,可以直接通过umi确定相同的特异性引物扩增获得的扩增子链中哪些是来源于突变或非突变,从而对突变进行定量检测,获得准确的突变率。
18.还需要说明的是,本技术的靶标特异性上游引物序列和靶标特异性下游引物序列
是针对人t细胞受体编码基因设计的全覆盖其cdr3区域编码基因序列的特异性引物序列,可以基于人基因组dna或者cdna快速捕获并扩增,覆盖人免疫组库tcr的cdr3功能区域,捕获效率高;并且,直接对pcr扩增富集获得的所有扩增子都添加相同唯一性标识符的产物进行测序文库构建和测序,即完成人免疫组库测序,减少了实验步骤,降低了dna损失,提高了最终结果的真实性;本技术的人免疫组库测序方法,可以高效整合免疫组库与illumina高通量测序平台的建库流程,利用本技术设计的引物,可实现两步pcr快速构建文库,整个实验时间可以缩短至4小时以内;此外,本技术的方法中,通过特殊设计引物,在免疫组库测序中加入umi,使得某一个模板核酸母链的所有扩增子链都添加相同的umi,能够校正每个靶点扩增性偏差,校正每个靶点扩增错误,实现靶标基因拷贝数定量检测,从而解决pcr扩增错误或人为因素导致的假阳性问题。
19.还需要说明的是,本技术的关键在于,引物结构的设计以及各引物的添加顺序的设计,使得最终扩增富集的扩增子中都带有相同的umi;至于具体的引物序列,可以根据具体针对的靶标序列和具体的测序平台而定。例如,采用常规的引物设计软件,针对具体的靶标序列设计第一引物的靶标特异性上游引物序列,针对预计采用的测序平台设计第一引物的测序平台上游引物结合区,由此组成第一引物。
20.本技术的一种实现方式中,靶标特异性上游引物序列为针对人t细胞受体β链的v基因设计的特异性引物序列;靶标特异性下游引物序列为针对人t细胞受体β链的j基因设计的特异性引物序列;通过靶标特异性上游引物序列和靶标特异性下游引物序列的扩增能够全覆盖t细胞受体β链的cdr3区域编码基因。
21.可以理解,t细胞受体β链的cdr3由v、d、j三个基因编码;因此,上游引物针对v基因设计,下游引物针对j基因设计,能够更好的确保全覆盖cdr3区域编码基因。
22.本技术的一种实现方式中,第一引物中,唯一性标识符的序列中插入有至少一个脱氧尿嘧啶,通过插入脱氧尿嘧啶的分隔,使得唯一性标识符的连续碱基数量小于5。
23.需要说明的是,本技术在引物中插入脱氧尿嘧啶或者利用脱氧尿嘧啶替换t,目的是为了在不需要的时候,利用udg/ung酶消化引物。在唯一性标识符中插入脱氧尿嘧啶,同样是为了尽量避免随机的umi可能在后续扩增中出现非特异性扩增,唯一性标识符序列中可以选择使用一个或几个固定的脱氧尿嘧啶插入至其碱基序列中间,脱氧尿嘧啶左右两边的连续性n碱基数量小于5nt,这样能够更有效的避免非特异性扩增;当然,如果不考虑非特异性扩增的可能性,唯一性标识符中也可以不插入脱氧尿嘧啶。
24.本技术的一种实现方式中,pcr扩增富集的扩增循环数大于或等于5。
25.需要说明的是,第三引物和第二引物的pcr扩增富集,主要是为了使最开始第一引物延伸标记umi的母链的扩增子能够被指数倍扩增富集,获得更多的来源于同一模板核酸的、且umi都相同的扩增子链,以便于后续建库和测序。
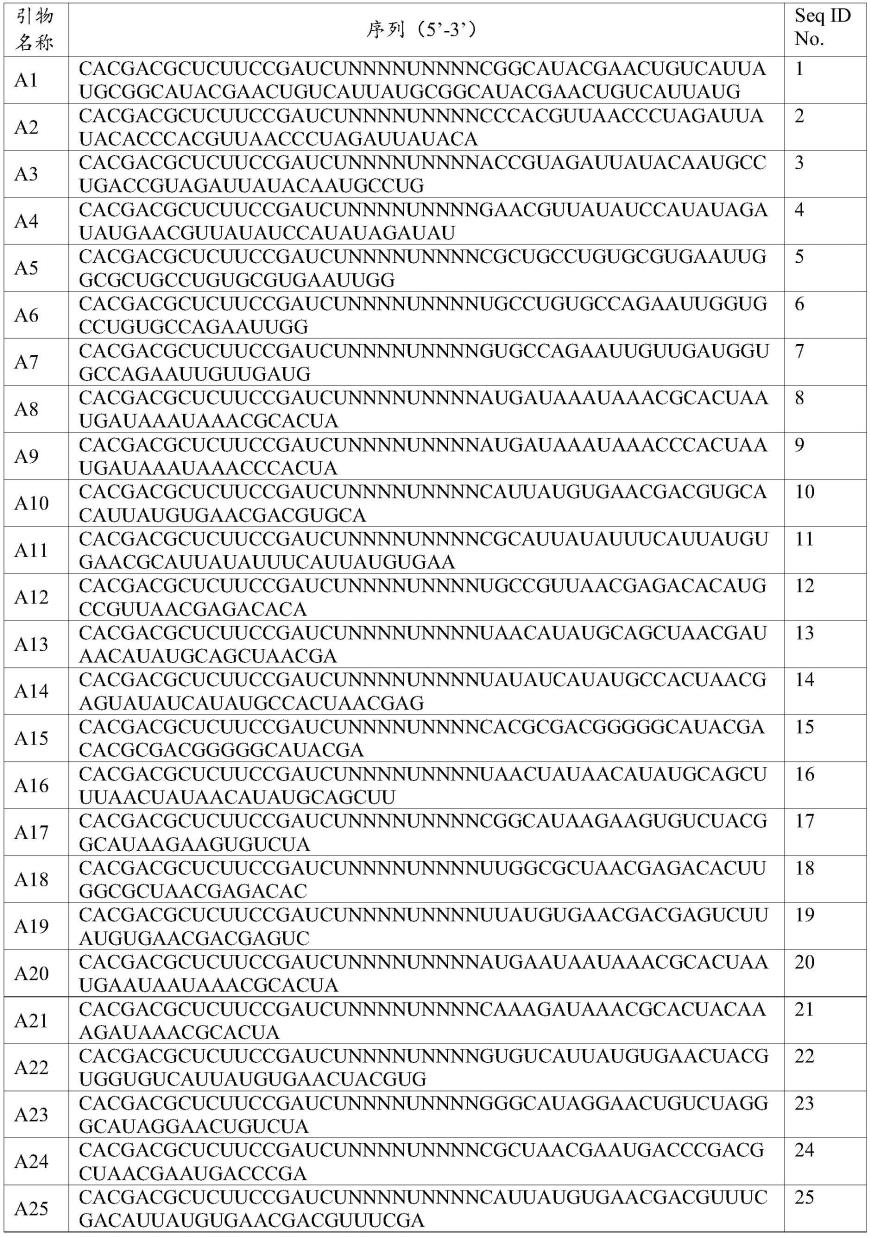
26.本技术的一种实现方式中,第一引物由seq id no.1至seq id no.40所示序列的40条引物组成。
27.需要说明的是,seq id no.1至seq id no.40所示序列的40条引物是本技术特别针对人类t淋巴细胞受体β链的v基因参考序列,根据碱基互补配对及引物设计原则,结合所需位点,自主设计的特异性上游引物;本技术设计的40条引物,包含了42个v功能区;使用时,40条引物按照摩尔质量等比混合即可。可以理解,本技术的40条引物只是本技术的一种
实现方式中证实具体可用的上游特异性引物,在本技术发明构思下,可用在本技术40条引物的基础上进行若干碱基的增减,或根据引物设计原则重新设计引物序列。
28.本技术的一种实现方式中,第二引物由seq id no.41至seq id no.52所示序列的12条引物组成。
29.需要说明的是,seq id no.41至seq id no.52所示序列的12条引物是本技术特别针对人类t淋巴细胞受体β链的j基因参考序列,根据碱基互补配对及引物设计原则,结合所需位点,自主设计的特异性下游引物;本技术的12条引物,包含6个j1功能区和7个j2功能区;使用时,12条引物按照摩尔质量等比混合即可。可以理解,本技术的12条引物只是本技术的一种实现方式中证实具体可用的下游特异性引物,在本技术发明构思下,可用在本技术12条引物的基础上进行若干碱基的增减,或根据引物设计原则重新设计引物序列。
30.本技术的一种实现方式中,第三引物为seq id no.53所示序列。
31.需要说明的是,本技术的第三引物实际上就是第一引物的测序平台上游引物结合区自5’端起的全部或部分序列,即针对测序平台的引物序列的3’末端设计的引物。例如,参考illumina novaseq6000测序平台的p5端3’末尾19nt序列设计的引物,即为seq id no.53所示序列。
32.本技术的一种实现方式中,测序文库构建包括以下步骤:
33.对pcr扩增富集获得的所有扩增子都添加相同唯一性标识符的产物进行纯化,获得纯化产物;采用第四引物和第五引物,对纯化产物进行建库扩增,获得测序文库;第四引物为带有测序接头和barcode的测序平台上游测序引物,第五引物为带有测序接头和barcode的测序平台下游测序引物。
34.需要说明的是,本技术的测序文库构建方法,实际上就是对第三引物和第二引物的pcr扩增富集产物进行扩增建库;即利用测序平台上游测序引物和测序平台下游测序引物对靶标产物进行再次扩增富集。
35.本技术的一种实现方式中,纯化为磁珠法纯化、过柱法纯化和凝胶法纯化中的至少一种。
36.本技术的一种实现方式中,第四引物为seq id no.54所示序列。
37.本技术的一种实现方式中,第五引物为seq id no.55所示序列。
38.需要说明的是,seq id no.54所示序列的第四引物和seq id no.55所示序列的第五引物中,“nnnnnn”是指长度为6-10nt的index,即barcode。例如,本技术的一种实现方式中,seq id no.54所示序列的第四引物的“nnnnnn”具体为“tgcgtaat”,seq id no.55所示序列的第五引物的“nnnnnn”具体为“cctaacct”。
39.本技术的另一面公开了一种人免疫组库测序的试剂盒,包括第一引物、第二引物、第三引物和udg/ung酶;第一引物由5’端到3’端依序包括测序平台上游引物结合区、唯一性标识符和靶标特异性上游引物序列;并且,第一引物中,测序平台上游引物结合区和靶标特异性上游引物序列中的碱基t替换为脱氧尿嘧啶,测序平台上游引物结合区对应测序平台的上游测序引物的3’末端;第二引物由5’端到3’端依序包括测序平台下游引物结合区和靶标特异性下游引物序列,测序平台下游引物结合区对应测序平台的下游测序引物的3’末端;第三引物为第一引物的测序平台上游引物结合区自5’端起的全部或部分序列,并且,第三引物中的碱基t不被替换为脱氧尿嘧啶;靶标特异性上游引物序列和所述靶标特异性下
游引物序列为针对人t细胞受体编码基因设计的全覆盖其cdr3区域编码基因序列的特异性引物序列。
40.需要说明的是,本技术的人免疫组库测序试剂盒,实际上就是将本技术人免疫组库测序的方法中使用的第一引物、第二引物、第三引物和udg/ung酶组装成试剂盒,以便于实现本技术的人免疫组库测序方法。因此,对试剂盒中第一引物、第二引物、第三引物的限定可以参考本技术的人免疫组库测序方法。例如,靶标特异性上游引物序列为针对人t细胞受体β链的v基因设计的特异性引物序列;靶标特异性下游引物序列为针对人t细胞受体β链的j基因设计的特异性引物序列;通过靶标特异性上游引物序列和靶标特异性下游引物序列的扩增能够全覆盖t细胞受体β链的cdr3区域编码基因。又例如,第一引物中,唯一性标识符的序列中插入有至少一个脱氧尿嘧啶,通过插入脱氧尿嘧啶的分隔,使得唯一性标识符的连续碱基数量小于5。
41.还需要说明的是,本技术的关键之一在于引物结构的设计,至于具体的引物序列,可以根据具体针对的靶标序列和具体的测序平台而定。例如,采用常规的引物设计软件,针对具体的靶标序列设计第一引物的靶标特异性上游引物序列,针对预计采用的测序平台设计第一引物的测序平台上游引物结合区,由此组成第一引物。
42.本技术的一种实现方式中,试剂盒中的第一引物由seq id no.1至seq id no.40所示序列的40条引物组成。
43.本技术的一种实现方式中,试剂盒中的第二引物由seq id no.41至seq id no.52所示序列的12条引物组成。
44.本技术的一种实现方式中,试剂盒中的第三引物为seq id no.53所示序列。
45.本技术的一种实现方式中,本技术的试剂盒还包括第四引物和第五引物;第四引物为带有测序接头和barcode的测序平台上游测序引物,第五引物为带有测序接头和barcode的测序平台下游测序引物。
46.需要说明的是,本技术的第四引物和第五引物实际上就是针对测序平台上游测序引物和测序平台下游测序引物设计,或者可以直接使用测序平台的上下游引物,只要其所带的测序接头和barcode与本技术相符即可。因此,第四引物和第五引物可以根据需求选择性的添加到试剂盒中。当然,为了使用方便,本技术的试剂盒中包含了第四引物和第五引物。
47.本技术的一种实现方式中,试剂盒中的第四引物为seq id no.54所示序列。
48.本技术的一种实现方式中,试剂盒中的第五引物为seq id no.55所示序列。
49.本技术的一种实现方式中,本技术的试剂盒还包括pcr扩增试剂。
50.可以理解,pcr扩增试剂可以根据需求组装到本技术的试剂盒中,也可以另行购买常规使用的pcr扩增试剂,例如pcr反应缓冲液、酶等。
51.由于采用以上技术方案,本技术的有益效果在于:
52.本技术的人免疫组库测序方法和试剂盒,对人t细胞受体进行特异性引物设计,全覆盖人免疫组库tcr的cdr3区域,捕获效率高;并且,通过对第一引物、第二引物和第三引物进行特殊设计,依序向反应体系中加入三种引物,使每个原始模板核酸只对应一个umi标签,从而校正每个靶点扩增性偏差,校正pcr扩增错误,校正建库过程中人为引入的扩增错误;本技术的方法和试剂盒能够对每个原始模板核酸进行标记,从而实现靶标基因的拷贝
数定量检测。
具体实施方式
53.本技术的人免疫组库测序方法基于ir-seq的多重pcr扩增法进行改进,以dna或cdna为模板进行扩增,例如,通过精心设计tcrβ链的cdr3上下游的v区、j区引物,特异性捕获富集人tcr cdr3区域,覆盖人tcrβ链的cdr3的42个功能区、2个d功能区、6个j1功能区、7个j2功能区。并在此基础上高度优化文库构建流程,减少了常规繁琐的实验操作,同时使用扩增子建库方法。同时在设计第二步扩增引物时,使用双端index标签设计扩增引物,保证数据的准确性。最重要的是,本技术创造性的采用特殊设计的第一引物、第二引物和第三引物,依序向反应体系中加入三种引物,给每个母链分子互补链插入唯一性umi标签,实现每个原始模板核酸只对应一个umi,校正每个靶点扩增性偏差,实现靶标基因拷贝数定量检测。
54.本技术的人免疫组库测序的方法,包括以下步骤:
55.配制反应体系,采用第一引物对模板核酸进行一次延伸,得到互补链;第一引物由5’端到3’端依序包括测序平台上游引物结合区、唯一性标识符和靶标特异性上游引物序列;并且,第一引物中,测序平台上游引物结合区和靶标特异性上游引物序列中的碱基t替换为脱氧尿嘧啶,测序平台上游引物结合区对应测序平台的上游测序引物的3’末端;
56.第一引物延伸结束后,向反应体系中加入第二引物,利用第二引物对第一引物延伸的互补链进行一次延伸,获得由测序平台上游引物结合区、唯一性标识符、靶标序列和测序平台下游引物结合区组成的产物;第二引物由5’端到3’端依序包括测序平台下游引物结合区和靶标特异性下游引物序列,测序平台下游引物结合区对应测序平台的下游测序引物的3’末端;
57.第二引物延伸结束后,向反应体系中加入udg/ung酶消化脱氧尿嘧啶,从而消化第一引物以及第一引物的延伸链;
58.udg/ung酶消化完成后,向反应体系中加入第三引物,利用第三引物和第二引物对第二引物延伸的产物进行pcr扩增富集,获得模板核酸的所有扩增子都添加相同唯一性标识符的产物;第三引物为第一引物的测序平台上游引物结合区自5’端起的全部或部分序列,并且,第三引物中的碱基t不被替换为脱氧尿嘧啶;
59.对pcr扩增富集获得的所有扩增子都添加相同唯一性标识符的产物进行测序文库构建和测序,即完成人免疫组库测序;
60.靶标特异性上游引物序列和靶标特异性下游引物序列为针对人t细胞受体编码基因设计的全覆盖其cdr3区域编码基因序列的特异性引物序列。
61.本技术的人免疫组库测序方法,其大致原理为:设计特异性umi序列,例如,结合人t细胞受体β链靶区域序列,延伸互补链同时加入umi,后利用udg/ung酶消化体系中未被使用的umi序列,实现每条模板分子的延伸链umi唯一性。再利用此互补链扩增富集靶向区域,设计相应的测序引物,给富集产物加入barcode/index及测序接头,完成建库。
62.本技术的人免疫组库测序方法通用性强,适合以dna为起始模板的tcr测序,或rna合成的cdna为起始模板进行多重pcr扩增,具有高特异性、高灵敏度特点。酶法消化多余umi标签,保证相同umi的分子链都来自于同一条模板,实现唯一性分子标签,可以矫正因pcr错
误或人为因素导致的假阳性问题,可以矫正多重pcr中扩增效率不均匀导致的偏差,保证数据真实性。
63.本技术的人免疫组库测序方法具有以下优点:
64.1.自主设计相关引物,可以基于人基因组dna或者cdna快速捕获并扩增,例如,覆盖人免疫组库tcrβ链cdr3功能区域,捕获效率高。
65.2.目前市场一些tcr产品使用琼脂糖凝胶电泳对捕获区域分选纯化,操作较为繁琐易错,且电泳法纯化容易造成核酸损失,导致实验结果失真。本技术的一种实现方式中,直接对pcr扩增富集获得的所有扩增子都添加相同唯一性标识符的产物进行纯化、测序文库构建和测序,例如使用特定比例ampure磁珠将dna纯化及片段分选一步化,减少实验步骤,降低dna损失,提高最终结果的真实性。
66.3.方法上,本技术高效整合人免疫组库与illumina高通量测序平台的建库流程,自主设计相关引物,可实现两步pcr快速构建文库;实验时间可以缩短至4小时以内。
67.4.特殊设计引物,并在免疫组库测序中加入umi,实现校正每个靶点扩增性偏差,校正每个靶点扩增错误,实现靶标基因拷贝数定量检测。
68.本技术方法中,第一引物、第二引物、第三引物、第四引物和第五引物的设计思路如下:
69.第一引物为umi序列,包括三个部分:第一部分为15-25nt的固定序列,对应测序平台上游测序引物3’末端,第二部分为6-8位随机性的n碱基序列,即umi,第三部分为靶标特异性上游引物序列。序列连接顺序为:5
’‑
第一部分-第二部分-第三部分-3’。
70.其中,在第一、三部分序列中,使用du(脱氧尿嘧啶)碱基替换t(胸腺嘧啶)碱基。第一部分15-25nt固定序列可参考不同测序平台的上游接头完整3’末端序列,例如参考illumina novaseq6000测序平台的p5端3’末尾19nt序列设计为seq id no.56所示序列,
71.seq id no.56:5
’‑
cacgacgcucuuccgaucu-3’。
72.进一步地,为了尽量避免随机的umi可能在后续扩增中出现非特异性扩增,第二部分序列中可以选择使用一个或几个固定的脱氧尿嘧啶插入至6-8位随机性的n碱基序列中间,脱氧尿嘧啶左右两边的连续性n碱基数量小于5nt,当然,如果不考虑非特异性扩增的可能性,第二部分序列可以不插入脱氧尿嘧啶。例如,第二部分序列可以是5
’‑
nnnnunnnn-3’,5
’‑
nnnunnnunn-3’或5
’‑
nnnnnnnn-3’等。
73.进一步地,第三部分序列为靶标特异性上游引物序列,可根据ncbi等权威数据库查找靶标基因,根据碱基互补配对及引物设计原则,结合所需位点,自主设计上游引物。可根据多靶点设计多个特异性强的pcr引物,混合使用。例如,本技术的一种实现方式中,参考ncbi、imgt标准数据库,查找人类t淋巴细胞受体β链的v基因参考序列;根据碱基互补配对及引物设计原则,结合所需位点,自主设计上游引物;本技术一共设计靶标特异性上游引物序列40种,共包含42个v功能区;将所有功能区引物按照摩尔质量等比混合使用。
74.第二引物,包括两个部分:第一部分为下游特异性引物序列,第二部分为15-25nt的固定序列,对应测序平台下游测序引物3’末端互补序列。序列连接顺序为:5
’‑
第二部分-第一部分-3’。
75.进一步地,第二引物的第一部分序列为靶标特异性下游引物序列,可根据ncbi等权威数据库查找靶标基因,根据碱基互补配对及引物设计原则,结合所需位点,自主设计下
游引物。可根据多靶点设计多个特异性强的pcr引物,混合使用。例如,本技术的一种实现方式中,参考ncbi、imgt标准数据库,查找人类t淋巴细胞受体β链的j基因参考序列;根据碱基互补配对及引物设计原则,结合所需位点,自主设计下游引物;本技术一共设计靶标特异性上游引物序列12种,共包含6个j1功能区和7个j2功能区;将共13个功能区引物按照摩尔质量等比混合使用。
76.第二引物的第二部分15-25nt固定序列可参考不同测序平台的下游接头完整3’末端序列,例如参考illumina novaseq6000测序平台的p7端3’末尾21nt序列的互补序列设计为seq id no.57所示序列,
77.seq id no.57:5
’‑
agacgtgtgctcttccgatct-3’。
78.第三引物为与第一引物序列中15-25nt的固定序列相同的序列,需要注意得的是,此段序列t碱基不可被脱氧尿嘧啶替换。例如参考illumina novaseq6000测序平台的p5端3’末尾19nt序列设计为seq id no.53所示序列,
79.seq id no.53:5
’‑
cacgacgctcttccgatct-3’。
80.第四引物的序列为:带有barcode的完整上游测序接头序列,index长度可以为6-10nt。例如参考illumina novaseq6000测序平台的p5序列设计为seq id no.54所示序列,
81.seq id no.54:
[0082]5’‑
aatgatacggcgaccaccgagatctacacnnnnnnacactctttccctacacgacgctcttccgatct-3’。
[0083]
seq id no.54所示序列的第四引物中“nnnnnn”即index序列。
[0084]
第五引物的序列为:带有barcode的下游测序接头序列的互补序列,index长度可以为6-10nt。例如参考illumina novaseq6000测序平台的p7序列设计为seq id no.55所示序列,seq id no.55:
[0085]5’‑
caagcagaagacggcatacgagatnnnnnngtgactggagttcagacgtgtgctcttccgatct-3’。
[0086]
seq id no.55所示序列的第五引物中“nnnnnn”即index序列。
[0087]
基于本技术的第一引物至第五引物的快速加入umi的方法及完整的文库制备技术,流程包括:1.使用第一引物延伸模板核酸,得到互补链,引入umi标签。2.加入第二引物完全延伸第1步中的互补链。3.使用udg/ung酶消化脱氧尿嘧啶,消化第一引物。4.加入第三引物对第2步产物进行扩增富集;并纯化扩增产物,去除体系、多余引物及基因组污染。5.使用第四引物及第五引物进行建库扩增,加上barcode及测序接头,完成建库。
[0088]
具体地,技术流程详细说明如下:
[0089]
第一步:引入umi标签,模板互补链延伸:
[0090]
取含靶区域的模板核酸,例如dna/cdna,总量1-100ng,优选地基因组dna为模板,优选地核酸量为100ng。
[0091]
配制延伸体系:可以使用市售pcr扩增试剂盒或者自研pcr扩增试剂盒,其主要组分可以包括但不限于:dna聚合酶、mg离子、dntps、缓冲体系。
[0092]
若实验设计为多重pcr反应,则优选选择市售或自研多重pcr扩增试剂盒。
[0093]
在配制好的延伸体系中,加入第一引物,其工作浓度为可以为50-500mm;优选地,工作浓度设置为200mm,即每条引物的浓度都是200mm。
[0094]
在配制好的延伸体系中,加入准备好的模板dna/cdna,充分混合后,进行模板互补链延伸反应。反应程序参数应参照pcr扩增试剂盒说明书进行设置。应注意的是,需将反应的延伸时间调整为大于“目标区域长度/延伸速度”,即使目标区域能够被充分、完整的延伸;不设置pcr循环数或者循环数设置为1个,即只进行一次延伸,不重复进行变形、退火、延伸。
[0095]
完成此反应后,得到的产物链即为已经加入umi的模板互补链。
[0096]
第二步:模板互补链互补延伸
[0097]
取第一步反应产物,加入第二引物,其工作浓度为可以为50-500mm;优选地,工作浓度为200mm,即每条引物的浓度都是200mm。
[0098]
混合均匀后,放入pcr程序,pcr程序与第一步保持完全一致。
[0099]
完成此反应后,得到的产物链即为已经加入umi的文库片段,且序列与模板上的目的片段一致。
[0100]
第三步:使用udg/ung酶消化脱氧尿嘧啶
[0101]
准备热敏性udg/ung酶,可以为市售或自制酶。
[0102]
取出第二步反应产物,加入准备好的热敏性udg/ung酶,酶加入量根据酶活力及消化效率调整,一般来说,酶活力大于1u/μl时,加入1μl即可。充分混匀后,根据酶最适反应温度及条件,将体系中所有含脱氧尿嘧啶的序列消化。
[0103]
此步的目的是为了消化体系中富余的第一引物以及参与第一步反应的第一引物延伸序列。最终,得到的产物中只含有初始加入的dna模板以及第二步产出的带有唯一性umi标签的延伸产物链。
[0104]
第四步:特异性扩增富集
[0105]
第三引物加入至混合的第三步产物中,其工作浓度为可以为50-500mm;优选地,工作浓度为200mm。
[0106]
充分混合后,进行模板互补链延伸反应。反应程序参数应参照pcr扩增试剂盒说明书进行设置。pcr循环数可以根据项目需求及试剂盒性能个性化设置,优选5个循环以上。
[0107]
扩增完成后,将产物全部取出,进行核酸纯化。得到纯度高无杂质的纯化产物,进行下一步建库扩增。纯化方法可以是但不限于磁珠法、过柱法或者凝胶法。
[0108]
第五步:建库扩增
[0109]
将第四步得到的纯化产物,加入扩增体系,及第四引物、第五引物,充分混合。第四引物、第五引物工作浓度可以为200-2000mm,优选1500mm。
[0110]
反应程序参数应参照pcr扩增试剂盒说明书进行设置。pcr循环数可以根据项目需求及试剂盒性能个性化设置。
[0111]
反应完成后,产物即为带有完整接头信息的可上机文库。通过核酸纯化,得到高纯度的文库。质检定量后,文库即可用于上机测序。
[0112]
下面通过具体实施例对本技术作进一步详细说明。以下实施例仅对本技术进行进一步说明,不应理解为对本技术的限制。
[0113]
实施例
[0114]
按照以上方法和思路本例设计了人免疫组库测序的第一引物至第五引物进行试验。本例在人免疫组库测序中加入umi矫正偏好性扩增及扩增错误,以基因组dna为模板,设
计40条上游v区引物,以及12条下游j区引物,覆盖该区所有亚型。本例设计的第一引物和第二引物对tcrβchain cdr3区域进行多重pcr扩增,消除偏好性扩增及扩增错误累积带来的影响,使结果更接近真实。具体如下:
[0115]
取一例从人外周血中提取的基因组dna样本备用。基因组dna样本由深圳市海普洛斯生物科技有限公司提供和保存。
[0116]
按照以上思路设计第一引物至第五引物:
[0117]
第一引物,本例参考ncbi、imgt标准数据库,查找人类t淋巴细胞受体β链的v基因参考序列;根据碱基互补配对及引物设计原则,结合所需位点,自主设计上游引物;本例一共设计靶标特异性上游引物序列40种,共包含42个v功能区;将所有功能区引物按照摩尔质量等比混合使用。本例的第一引物由seq id no.1至seq id no.40所示序列的40条引物组成,如表1所示。
[0118]
第二引物,本例参考ncbi、imgt等标准数据库,查找人类t淋巴细胞受体β链的j基因参考序列;根据碱基互补配对及引物设计原则,结合所需位点,自主设计下游引物;本例一共设计靶标特异性上游引物序列12种,共包含6个j1功能区和7个j2功能区;将所有功能区引物按照摩尔质量等比混合使用。本例的第二引物由seq id no.41至seq id no.52所示序列的12条引物组成,如表1所示。
[0119]
表1第一引物和第二引物
[0120]
3’;
[0131]
seq id no.55所示序列的第五引物中,“nnnnnn”具体为“cctaacct”。
[0132]
设计合成以上引物后,用te buffer将引物稀释,第一引物、第二引物、第三引物稀释至浓度5μm,第四引物、第五引物稀释至浓度30μm。
[0133]
取100ng上述该例样本基因组dna,使用qiagen multiplexpcr试剂盒进行扩增实验。
[0134]
配制反应体系,依次加入到一个新的0.2ml管中,反应体系为:第一引物4.5μl、基因组dna100ng、pcr master mix 25μl、q-solution 5μl,补充nf水至45μl。
[0135]
轻轻混匀混有样本及试剂的0.2ml样本管后,放入biorad t100pcr仪上进行pcr反应,反应程序:95℃变性15min,然后94℃30s、60℃90s、72℃90s,最后72℃延伸5min,4℃待机。
[0136]
反应完成后,取出反应产物,加入5μl第二引物,混匀后放入biorad t100pcr仪上进行pcr反应,反应程序:95℃变性15min,然后94℃30s、60℃90s、72℃90s,最后72℃延伸5min,4℃待机。
[0137]
反应完成后,取出反应产物,加入1μl热敏性udg酶(heat-labile udg,vazyme),混匀后放入以下程序中反应:25℃消化10min,55℃灭活5min,95℃5min,4℃待机。
[0138]
反应完成后,取出反应产物,加入5μl第三引物,混匀后放入biorad t100pcr仪上进行pcr反应,反应程序:95℃变性15min,然后进入30个循环:94℃30s、56℃90s、72℃90s,循环结束后72℃延伸5min,4℃待机。
[0139]
反应完成后,取出反应产物,使用磁珠纯化,详细步骤如下:
[0140]
1.使用1.2
×
ampure xp beads磁珠进行多重pcr产物纯化:取新1.5ml样本管,将多重pcr产物50μl与60μl混合均匀的ampure xp beads磁珠加入新1.5ml样本管中,涡旋混匀,置于室温10min,使dna与磁珠充分结合。将1.5ml样本管置于磁力架上,进行磁珠吸附,直至溶液澄清,小心移除上清液。
[0141]
2.再加入500μl的80%乙醇,180度旋转样本管使磁珠穿过溶液吸至另一侧管壁,旋转2-3次,静置15s后弃上清液。
[0142]
3.重复步骤2一次;
[0143]
4.自然静置1.5ml样本管,待酒精全部挥发后,向1.5ml样本管中加入20μl无核酸酶水,充分混匀。将1.5ml样本管置于磁力架上,进行磁珠吸附,直至溶液澄清,小心吸出上清液放入新0.2μl样本管中,得到纯化产物。
[0144]
测序文库构建,将第四引物、第五引物、扩增试剂与纯化产物混合,进行建库扩增。使用kapa hifi hotstart ready mix试剂进行扩增,混合比例如下:2
×
kapa hifi hotstart readymix 25μl、第四引物2.5μl、第五引物2.5μl、纯化产物20μl,补充nf水至50μl。
[0145]
混合均匀后,放入以下程序中反应:98℃变性45s,然后进入5个循环:98℃15s、60℃30s、72℃30s,循环结束后72℃延伸1min,4℃待机。
[0146]
程序结束后,得到建库扩增pcr产物50μl,使用1
×
ampure xp磁珠进行多重pcr产物纯化:
[0147]
取新1.5ml样本管,将多重pcr产物50μl与50μl混合均匀的ampure xp磁珠加入新
1.5ml样本管中,涡旋混匀,置于室温10min,使dna与磁珠充分结合。将1.5ml样本管置于磁力架上,进行磁珠吸附,直至溶液澄清,小心移除上清液。
[0148]
加入500μl的80%乙醇,180度旋转样本管使磁珠穿过溶液吸至另一侧管壁,旋转2-3次,静置15s后弃上清液;重复此步骤一次。
[0149]
自然静置1.5ml样本管,待酒精全部挥发后,向1.5ml样本管中加入20μlnuclease-freewater,充分混匀。将1.5ml样本管置于磁力架上,进行磁珠吸附,直至溶液澄清,小心吸出上清液,标记保存,即完成扩增子文库建库。
[0150]
高通量测序:使用illumina novaseq 6000高通量测序系统进行上机测序,上机模式为pe151+8+8+151。
[0151]
结果:
[0152]
对测序获得的下机数据进行常规的质控过滤和测序深度阈值过滤后,分析序列umi。本例分为两种方法进行分析,第一种分析方法:作为对比分析数据,不考虑umi的存在,不过滤umi序列直接分析上游v区及下游j区的各功能区占比、分析多样性、克隆指数及相关指标。第二种分析方法:过滤umi序列,umi与cdr3序列一致的序列认为来源于同一母链模板,算为一个拷贝数,在此之上,对比相同umi的所有cdr3序列,分析其read是否存在单个或几个偶发性的碱基差异,如有则过滤该差异序列,矫正扩增及实验引入的错误,以此分析上游v区及下游j区的各功能区占比、分析多样性、克隆指数及相关指标。分析结果如表2所示。
[0153]
表2对比分析umi序列对人免疫组库测序结果的影响
[0154]
分析方法不过滤umi过滤umi总reads数(条)1250345212503452错误碱基占比(%)9898总克隆reads(条)65593786559378抓取reads占比(%)9898未抓取reads(%)22随机抓取raw_read(条)10000001000000唯一克隆氨基酸序列数(条)3045620903最高频率氨基酸序列占比(%)0.130.04香农指数11.210.2
[0155]
表2中,“不过滤umi”即第一种分析方法,“过滤umi”即第二种分析方法,“总reads数”是指原始数据中包含的总reads数,“错误碱基占比”是指错误率小于0.1%的碱基占比,“抓取reads占比”是指抓取到cdr3区域reads占比,“未抓取reads”是指未抓取到的cdr3区域reads的占比,“随机抓取raw_read”是指随机抓取raw_read用于分析的read条,“唯一克隆氨基酸序列数”是指唯一的克隆氨基酸序列数,“香农指数”值越高,免疫组库多样性越高。
[0156]
结果显示,在不过滤umi的情况下,由于偏好性扩增无法去除,会导致最高频率氨基酸序列占比虚高,使用umi过滤,则可以去除偏好性扩增,更加真实地反映最高cdr3占比。另外,在不过滤umi的数据下,唯一的克隆氨基酸序列数与香农指数相比更高,原因是在高扩增循环数下,累积了大量扩增错误,在无umi情况下很难识别这些序列是模板本身序列还是实验引入错误,然而,错误的序列也会增加cdr3的唯一性和多样性,进入到分析环节,从
而造成指标虚高。使用umi过滤的数据则更接近真实情况,矫正扩增偏差带来的不准确定量、矫正因扩增错误或实验引入的错误导致的分析指标虚高,提高了数据可信度。
[0157]
因此,本例的人免疫组库测序方法在测序错误中有良好的矫正作用,可以矫正扩增偏差带来的不准确定量、矫正因扩增错误或实验引入的错误导致的阳性率虚高/假阳性。
[0158]
以上内容是结合具体的实施方式对本技术所作的进一步详细说明,不能认定本技术的具体实施只局限于这些说明。对于本技术所属技术领域的普通技术人员来说,在不脱离本技术构思的前提下,还可以做出若干简单推演或替换。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1