电致变色化合物及其制备方法、阴极材料和电致变色器件与流程
1.本发明涉及光电材料技术领域,具体而言,涉及一种电致变色化合物及其制备方法、阴极材料和电致变色器件。
背景技术:
2.电致变色是指材料的光学属性在外加电场的作用下发生稳定且可逆的颜色变化的现象,在外观上表现为颜色和透明度的可逆变化,具有电致变色能力的材料被称为电致变色材料,使用电致变色材料制造的器件称为电致变色器件,电致变色器件的性能与其中的电致变色材料的性能关系密切,合适的电致变色材料的选择能赋予电致变色器件颜色变化可控、颜色变化范围大、双稳态、低驱动电压和使用寿命长等优点,电致变色器件在智能窗、防眩目后视镜以及显示器等领域具有广阔的应用前景。
3.紫精又名紫罗精,是典型的有机小分子电致变色材料,在电压驱动下,紫精类化合物具有良好的可逆氧化还原反应性能,反应同时伴随着显著的颜色变化,紫精类化合物具有变色对比度高、变色响应时间短、制备成本低、使用寿命长等优点,属于已经被广泛研究的电致变色化合物。但是现有紫精类化合物组成的器件稳定性能较差,长时间和多次着/褪色循环后,老化严重,性能有很大的衰减,例如变色时间,不同状态下透光率差值等参数比使用之初均有变化。
4.有鉴于此,特提出本发明。
技术实现要素:
5.本发明的主要目的在于提供一种电致变色化合物及其制备方法、阴极材料和电致变色器件,以改善现有紫精类化合物组成的器件稳定性较差,长时间和多次着/褪色循环后,产品老化严重,性能有很大衰减的技术问题。
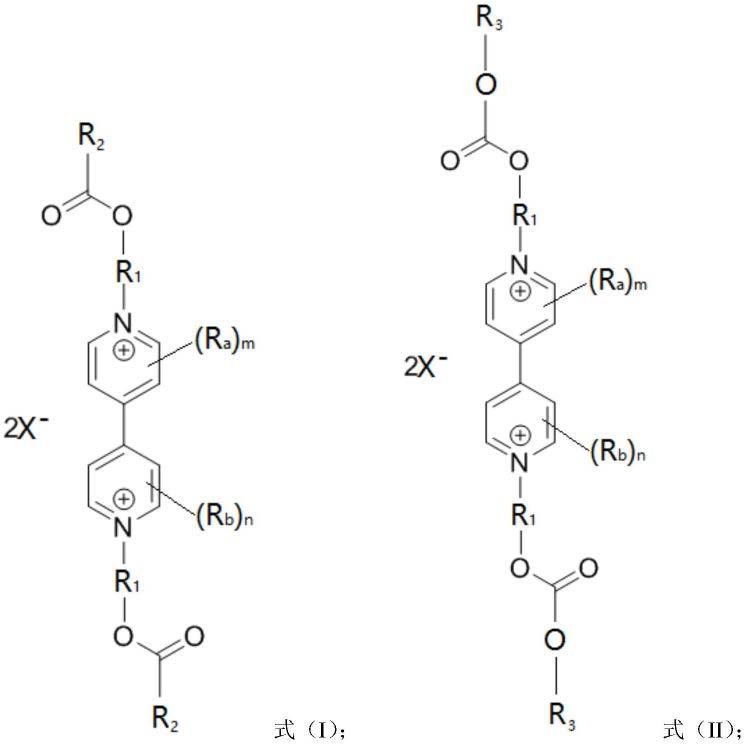
6.为了实现上述目的,根据本发明的一个方面,提供了一种电致变色化合物,该电致变色化合物具有如下式(i)或式(ii)所示结构:
[0007][0008]
r1表示c1~c12的直链或支链亚烷基;r2和r3各自独立地表示c1~c12的直链或支链烷基;ra和rb各自独立地表示氢、卤素、c1~c10的直链或支链烷基、-no2、-cn、-ora、-sra,其中ra表示以下第一基团:氢、c1~c10的直链或支链烷基、c3~c10的环烷基、c4~c10的环烷基烷基;m和n各自独立地表示0~4之间的整数,且当m大于或等于2的情况下,各ra可以相同也可以不同,当n大于或等于2的情况下,各rb可以相同也可以不同;x-表示cl-、br-、i-、clo
4-、ch3coo-、pf
6-、asf
6-、bf
4-、tfsi-、no
3-,且2个x-可以相同也可以不同。
[0009]
进一步地,r1表示c1~c6的直链或支链亚烷基;和/或,r2和r3各自独立地表示c1~c6的直链或支链烷基;和/或,ra和rb各自独立地表示氢、卤素、c1~c6的直链或支链烷基、-no2、-cn、-ora、-sra,其中,ra表示以下第一基团:氢、c1~c6的直链或支链烷基;和/或,x-表示clo
4-、pf
6-、bf
4-、tfsi-。
[0010]
进一步地,r1表示亚甲基、亚乙基、亚正丙基、亚异丙基、亚正丁基、亚异丁基;和/或,r2和r3各自独立地表示甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基;和/或,ra和rb各自独立地表示氢、卤素、甲基、乙基、-no2、-cn、-oh、-och3、-och2ch3。
[0011]
进一步地,上述电致变色化合物选自如下化合物中的至少一种:
[0012][0013]
为了实现上述目的,根据本发明的另一个方面,提供了一种电致变色化合物的制备方法,该制备方法包括:
[0014]
步骤s1,将卤代酸酯类化合物或卤代碳酸酯类化合物与4,4'-联吡啶类化合物混合进行取代反应,得到第一产物体系,将第一产物体系进行一次提纯,得到卤化电致变色化合物;以及可选的步骤s2,将卤代电致变色化合物与阴离子为x-的盐混合进行置换反应,得到第二产物体系,将第二产物体系进行二次提纯,得到电致变色化合物;
[0015]
其中,卤代酸酯类化合物具有式(ⅲ)所示结构,卤代碳酸酯类化合物具有式(iv)所示结构,4,4'-联吡啶类化合物具有式(v)所示结构;
[0016][0017]
上述z表示卤素,上述x-、r1、r2、r3、ra、rb、m、n均具有上述第一方面提供的电致变色化合物中的相同含义。
[0018]
进一步地,上述步骤s1,卤代酸酯类化合物或卤代碳酸酯类化合物与4,4'-联吡啶类化合物在第一溶剂中进行取代反应,取代反应的温度为65-100℃,取代反应的时间为60-100h。
[0019]
进一步地,上述一次提纯的方式包括依次进行的固液分离、洗涤以及干燥。
[0020]
进一步地,第一溶剂包括乙腈、乙醇、乙二醇、二甲基亚砜、二甲基甲酰胺、甲苯、乙酸乙酯、三氯甲烷、丙酮、四氢呋喃、1,4-二氧六环或水中的至少一种。
[0021]
进一步地,上述步骤s2,卤化电致变色化合物与阴离子为x-的盐在第二溶剂中进
行置换反应,该置换反应的温度为15-30℃,置换反应的时间为3-6h。
[0022]
进一步地,二次提纯的方式包括依次进行的固液分离、洗涤、重结晶和干燥。
[0023]
进一步地,阴离子为x-的盐包括铵盐、四乙基胺盐或四丁基胺盐中的至少一种。
[0024]
进一步地,第二溶剂包括水、乙腈、乙醇、乙二醇、二甲基亚砜、二甲基甲酰胺、甲苯、乙酸乙酯、三氯甲烷、丙酮、四氢呋喃或1,4-二氧六环中的至少一种。
[0025]
根据本发明的第三个方面,还提供了一种阴极材料,该阴极材料包括上述第一方面提供的任一种电致变色化合物或根据上述第二方面提供的任一种制备方法得到的电致变色化合物。
[0026]
根据本发明的第四个方面,还提供了一种电致变色器件,该电致变色器件包括上述第一方面提供的任一种电致变色化合物、上述第二方面提供的任一种制备方法制备得到的电致变色化合物或上述第三方面提供的任一种阴极材料。
[0027]
根据本发明的第五个方面,还提供了一种电致变色器件,该电致变色器件包括液态器件、凝胶器件或固态器件中的至少一种。
[0028]
应用本发明的技术方案,具备上述式(i)或式(ii)所示结构的电致变色化合物组成的电致变色器件具备优异的循环稳定性,长时间多次着/褪色循环后性能无明显衰减,同时还具备变色速度快,不同状态下透光率差值高的特性,有效延长了使用寿命。
附图说明
[0029]
构成本技术的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
[0030]
图1示出了本发明实施例1提供的电致变色化合物的核磁谱图以及
[0031]
图2(a)为实施例1提供电致变色化合物组成的电致变色器件的初始着色态和初始褪色态的光谱图;图2(b)为实施例1提供的电致变色化合物组成的电致变色器件循环20000次后的着色态和褪色态光谱图。
具体实施方式
[0032]
需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
[0033]
如本技术背景技术所分析的,现有紫精类化合物组成的器件存在稳定性较差,长时间和多次着/褪色循环后,老化严重,性能有很大衰减的技术问题。为了改善该问题,本技术提供了一种电致变色化合物及其制备方法、阴极材料、电致变色器件及其应用。
[0034]
在本技术的一种典型实施方式中,提供了一种电致变色化合物,该电致变色化合物具有如下式(i)或式(ii)所示结构:
[0035][0036]
上述r1表示c1~c12的直链或支链亚烷基;r2和r3各自独立地表示c1~c12的直链或支链烷基;ra和rb各自独立地表示氢、卤素、c1~c10的直链或支链烷基、-no2、-cn、-ora、-sra,其中ra表示以下第一基团:氢、c1~c10的直链或支链烷基、c3~c10的环烷基、c4~c10的环烷基烷基;m和n各自独立地表示0~4之间的整数,且当m大于或等于2的情况下,各ra可以相同也可以不同,当n大于或等于2的情况下,各rb可以相同也可以不同;x-表示cl-、br-、i-、clo
4-、ch3coo-、pf
6-、asf
6-、bf
4-、tfsi-、no
3-,且2个x-可以相同也可以不同。
[0037]
上述烷基包括取代烷基和非取代烷基,上述亚烷基包括取代亚烷基和非取代亚烷基,取代基为本领域常用取代基团,如:氯、溴、碘、-oh、-cn、-no2、-och3等,在此不再赘述。上述卤素包括但不限于氯、溴或碘等。上述tfsi-指的是双三氟甲基磺酰亚胺阴离子。
[0038]
需要说明是,上述式(i)和式(ii)中的r1可以相同也可以不同。
[0039]
应用本发明的技术方案,具备上述式(i)或式(ii)所示结构的电致变色化合物组成的电致变色器件具备优异的循环稳定性,长时间多次着/褪色循环后性能无明显衰减,同时还具备变色速度快,不同状态下透光率差值高的特性,有效延长了使用寿命。
[0040]
在本技术的一些实施例中,r1表示c1~c6的直链或支链亚烷基;r2和r3各自独立地表示c1~c6的直链或支链烷基;ra和rb各自独立地表示氢、卤素、c1~c6的直链或支链烷基、-no2、-cn、-ora、-sra,其中ra表示以下第一基团:氢、c1~c6的直链或支链烷基;x-表示clo
4-、pf
6-、bf
4-、tfsi-时,上述电致变色化合物组成的电致变色器件具备更为优异的循环稳定性。尤其是当r1表示亚甲基、亚乙基、亚正丙基、亚异丙基、亚正丁基、亚异丁基;r2和r3各自独立地表示甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基;ra和rb各自独立地表示氢、卤素、甲基、乙基、-no2、-cn、-oh、-och3、-och2ch3时,上述电致变色化合物组成
的电致变色器件的循环稳定性更佳。
[0041]
在本技术的一些实施例中,电致变色化合物选自如下化合物中的至少一种时,其组成的电致变色器件的循环稳定性更为显著,长时间和多次着/褪色循环后,变色时间以及不同状态下透光率差值等参数比使用之初无明显差异,使用寿命进一步延长。
[0042][0043]
在本技术的另一种典型实施方式中,还提供了一种电致变色化合物的制备方法,该制备方法包括:步骤s1,将卤代酸酯类化合物或卤代碳酸酯类化合物与4,4'-联吡啶类化合物混合进行取代反应,得到第一产物体系,将第一产物体系进行一次提纯,得到卤化电致变色化合物;其中,卤代酸酯类化合物具有式(ⅲ)所示结构,卤代碳酸酯类化合物具有式(iv)所示结构,4,4'-联吡啶类化合物具有式(v)所示结构;
[0044][0045]
上述z表示卤素,x-、r1、r2、r3、ra、rb、m、n均具有上述第一种典型实施方式提供的任一种电致变色化合物中的相同含义。
[0046]
上述卤化电致变色化合物指的是具有上述式(i)或式(ii)所示结构,且x-为卤素离子的电致变色化合物。上述卤代酸酯类化合物与4,4'-联啶类化合物混合进行取代反应,制备得到的是具有上述式(i)所示结构,且x-为卤素离子的电致变色化合物;上述卤代碳酸酯类化合物与4,4'-联啶类化合物混合进行取代反应,制备得到的是具有上述式(ii)所示结构,且x-为卤素离子的电致变色化合物。
[0047]
本技术提供的电致变色化合物的制备方法工艺简单,操作简便,适用于规模化大
生产,能够有效提高制备效率。
[0048]
为了进一步提高上述取代反应的效率,促进上述取代反应进行的更加充分,优选上述步骤s1,卤代酸酯类化合物或卤代碳酸酯类化合物与4,4'-联吡啶类化合物在第一溶剂中进行取代反应,取代反应的温度为65-100℃,取代反应的时间为60-100h。
[0049]
上述第一溶剂的类型不作限制,任何能够促进4,4'-联吡啶类化合物与卤代酸酯类化合物或卤代碳酸酯类化合物溶解或分散的溶剂类型均可,包括但不限于乙腈、乙醇、乙二醇、二甲基亚砜、二甲基甲酰胺、甲苯、乙酸乙酯、三氯甲烷、丙酮、四氢呋喃、1,4-二氧六环或水中的任意一种或多种形成的混合溶剂。
[0050]
上述一次提纯的方式也不作限制,任何能够将卤化电致变色化合物提纯的方式均可,包括但不限于固液分离、结晶、洗涤等方式。在本技术的一些实施例中,一次提纯包括依次进行的固液分离、洗涤和干燥。该固液分离的方式包括但不限于离心、过滤等。上述洗涤所采用的溶剂类型不作限制,任何与卤代电致变色化合物不相溶的溶剂均可,为了进一步提高卤化电致变色化合物表面的提纯效率,洗涤采用的溶剂包括但不限于乙腈、丙酮等。上述干燥的方式包括但不限于风干、烘干、滤干等,为了提高干燥效率,优选为滤干。
[0051]
典型但非限制性的,上述取代反应的温度如为65℃、70℃、75℃、80℃、85℃、88℃、90℃、92℃、95℃、98℃或100℃,取代反应的时间如为60h、65h、70h、75h、80h、85h、90h、92h、95h、98h或100h。
[0052]
由于上述卤化电致变色化合物中的阴离子为卤素离子,某些卤素离子如溴离子等,易于与其它物质发生反应,导致卤化电致变色化合物不稳定,为了进一步提高电致变色化合物的稳定性,优选上述电致变色化合物的制备方法还包括步骤s2,将上述卤化电致变色化合物与阴离子为x-的盐混合进行置换反应,得到第二产物体系,将第二产物体系进行二次提纯,得到电致变色化合物。
[0053]
为了进一步提高上述置换反应的效率,优选上述步骤s2,卤化电致变色化合物与阴离子为x-的盐在第二溶剂中进行置换反应,置换反应的温度为15-30℃,置换反应的时间为3-6h。
[0054]
上述阴离子为x-的盐的类型不作限制,任何能够与卤化电致变色化合物进行阴离子置换的盐的类型均可,包括但不限于铵盐、四乙基胺盐或四丁基胺盐中的任意一种或多种的混合物。
[0055]
上述第二溶剂的类型不作限制,任何能够促进卤化电致变色化合物与阴离子为x-的盐进行阴离子置换的溶剂类型均可,从降低成本以及环保的角度出发,优选为水。
[0056]
上述二次提纯的方式也不作限制,任何能够将电致变色化合物提纯的方式均可,包括但不限于固液分离、结晶、洗涤等方式。在本技术的一些实施例中,二次提纯包括依次进行的固液分离、洗涤、重结晶和干燥。该固液分离的方式包括但不限于离心、过滤等。上述洗涤所采用的溶剂类型不作限制,任何与电致变色化合物不相溶的溶剂均可,从环保以及降低成本的角度出发,洗涤采用的溶剂优选为水等。上述重结晶所采用的溶剂类型不作限制,任何能够将电致变色化合物进行重结晶的溶剂均可,包括但不限于乙腈和/或甲醇等。上述干燥的方式包括但不限于风干、烘干、滤干等,为了提高干燥效率,优选为滤干。
[0057]
在本技术的第三种典型实施方式中,提供了一种阴极材料,该阴极材料包括上述第一种典型实施方式中提供的任一种电致变色化合物或根据第二种典型实施方式提供的
任一种制备方法得到的电致变色化合物。
[0058]
应用本发明的技术方案,包含具备上述式(i)或式(ii)所示结构的电致变色化合物的阴极材料组成的电致变色器件具备优异的循环稳定性,长时间多次着/褪色循环后性能无明显衰减,同时还具备变色速度快,不同状态下透光率差值高的特性,有效延长了使用寿命。
[0059]
在本技术的第四种典型实施方式中,提供了一种电致变色器件,该电致变色器件包括液态器件,凝胶器件或固态器件中的任意一种或多种不同形态器件的组合。
[0060]
应用本发明的技术方案,由上述式(i)或式(ii)所示结构的电致变色化合物组成的电致变色器件具备优异的循环稳定性,长时间多次着/褪色循环后性能无明显衰减,同时还具备变色速度快,不同状态下透光率差值高的特性,有效延长了使用寿命。
[0061]
下面将结合实施例和对比例,进一步说明本技术的有益效果。
[0062]
实施例1
[0063]
本技术提供了一种电致变色化合物,其结构式如下所示:
[0064][0065]
其制备方法按照以下步骤进行:
[0066]
(1)将4,4'-联吡啶(50g,0.32mol)和2-溴乙基乙酸酯(534g,3.2mol)溶解在乙腈(500ml)中,加热至90℃,反应96小时,得到第一产物体系;将第一产物体系趁热过滤得到滤饼,用乙腈和丙酮冲洗滤饼,抽干,得到溴化电致变色化合物;
[0067]
(2)将上述溴化电致变色化合物和六氟磷酸铵(208.72g,1.3mol)溶解于水中,室温反应4小时,得到第二产物体系;将第二产物体系抽滤得到滤饼,将滤饼用水冲洗后,再将滤饼分散在乙腈和甲醇的混合溶液(乙腈和甲醇的体积比为1:2)中进行重结晶,然后抽干,得到上述电致变色化合物(109.3g,收率55%)。
[0068]
上述电致变色化合物制备反应式如下所示:
[0069][0070]
实施例2
[0071]
本技术提供了一种电致变色化合物,其结构式如下所示:
[0072][0073]
其制备方法按照以下步骤进行:
[0074]
(1)将4,4'-联吡啶(50g,0.32mol)和2-溴乙基乙基碳酸酯(627g,3.2mol)溶解在乙腈(500ml)中,加热至90℃,反应96小时,得到第一产物体系;将第一产物体系趁热过滤得到滤饼,用乙腈和丙酮冲洗滤饼,抽干,得到溴化电致变色化合物;
[0075]
(2)将上述溴化电致变色化合物和六氟磷酸铵(208.72g,1.3mol)溶解于水中,室温反应4小时,得到第二产物体系;将第二产物体系抽滤得到滤饼,将滤饼用水冲洗后,再将滤饼分散在乙腈和甲醇的混合溶液(乙腈和甲醇的体积比为1:2)中进行重结晶,然后抽干,得到上述电致变色化合物(100g,收率46%)。
[0076]
上述电致变色化合物制备反应式如下所示:
[0077][0078]
实施例3
[0079]
本实施例提供了一种电致变色化合物,其结构式如下所示:
[0080][0081]
其制备方法与实施例1的不同之处在于,步骤(2)中,采用1.3mol的高氯酸铵
(nh4clo4)替换六氟磷酸铵,得到的电致变色化合物的质量为57g,收率为34%。
[0082]
实施例4
[0083]
本实施例提供了一种电致变色化合物,其结构式如下所示:
[0084][0085]
其制备方法与实施例2的不同之处在于,步骤(2)中,采用1.3mol的高氯酸铵(nh4clo4)替换六氟磷酸铵,得到的电致变色化合物的质量为51g,收率为31%。
[0086]
实施例5
[0087]
本实施例提供了一种电致变色化合物,其结构式如下所示:
[0088][0089]
其制备方法与实施例1的不同之处在于,步骤(2)中,采用1.3mol的四氟硼酸铵(nh4bf4)替换六氟磷酸铵,得到的电致变色化合物的质量为63g,收率为39%。
[0090]
实施例6
[0091]
本实施例提供了一种电致变色化合物,其结构式如下所示:
[0092][0093]
其制备方法与实施例1的不同之处在于,步骤(2)中,采用1.3mol的四氟硼酸铵(nh4bf4)替换六氟磷酸铵,得到的电致变色化合物的质量为63g,收率为35%。
[0094]
实施例7
[0095]
本实施例提供了一种电致变色化合物,其结构式如下所示:
[0096][0097]
其制备方法同实施例1中步骤(1),得到的溴化电致变色化合物的质量为110g,收率为70%。
[0098]
实施例8
[0099]
本实施例提供了一种电致变色化合物,其结构式如下所示:
[0100][0101]
其制备方法同实施例2中步骤(1),得到的溴化电致变色化合物的质量为107g,收率为61%。
[0102]
对比例1
[0103]
本对比例提供了一种电致变色化合物,其具有如下结构:
[0104][0105]
对比例2
[0106]
本实施例提供了一种电致变色化合物,其具有如下结构:
[0107][0108]
试验例1
[0109]
将实施例1和实施例2进行核磁测试,图1为实施例1提供的电致变色化合物的核磁谱图;如图1所示,实施例1提供的电致变色化合物的1hnmr(d6-dmso,500mhz)谱图中σ(ppm)分别为:9.40(d,4h),8.81(d,4h),4.99(m,4h),4.60(m,4h),2.01(s,6h)。
[0110]
实施例2提供的电致变色化合物的1hnmr(d6-dmso,500mhz)谱图中σ(ppm)分别为:9.42(d,4h),8.78(d,4h),4.90(m,4h),4.60(m,4h),4.22(q,4h),1.32(t,6h)。
[0111]
试验例2
[0112]
取2片40mm
×
45mm的ito导电玻璃,用框胶将2片玻璃的ito面以面对面的的方式贴合起来,形成100μm高的空腔,并留有一个灌注孔:从灌注孔向空腔中灌入电致变色溶液,然后用uv胶封口,制得电致变色器件;其中,该电致变色溶液由5,10-二甲基吩嗪、实施例1提供的电致变色化合物和溶剂1,4-丁内酯组成,其中,5,10-二甲基吩嗪和电致变色化合物的摩尔浓度均各自独立地为85mmol/l。
[0113]
分别测试上述电致变色器件在不同波长照射下初始着色态(施加0.9v电压)、初始
褪色态(施加0v电压)、循环20000次后的着色态(施加0.9v电压)以及循环20000次后的褪色态(施加0v电压时)的透光率,并绘制相应的波长-透光率光谱图。其中上述1次循环指的是以信号发生器为电源,设置信号发生器的循环电压高电压为0.9v,低电压为0v,循环周期为30s,高电压与低电压持续时间均为15s。
[0114]
图2(a)为实施例1提供电致变色化合物组成的电致变色器件的初始着色态和初始褪色态的光谱图;图2(b)为实施例1提供的电致变色化合物组成的电致变色器件循环20000次后的着色态和褪色态光谱图。通过对比图2(a)和图2(b)可以看出,初始态着色态光谱与循环20000次后的着色态光谱基本吻合,初始态褪色态光谱与循环20000次后的褪色态光谱基本吻合,这说明上述电致变色器件,经过20000次着/褪色循环后性能无明显衰减,具有优异的循环稳定性。此外,通过上述图2(a)和图2(b)还可以看出,上述电致变色器件在相同波长下具备透光率差值高的特性。
[0115]
试验例3
[0116]
按照上述试验例2的方法分别制备由实施例1-6以及对比例1-2提供的电致变色化合物组成的电致变色器件。分别测试上述电致变色器件的初始以及20000次着褪色循环之后的褪色态透过率、着色态透过率、着色时间、褪色时间,结果如下表1所示。
[0117]
其中,上述初始褪色态透过率、初始着色态透过率、循环20000次后褪色态透过率、循环20000次后着色态透过率分别按照gb/t268094《建筑玻璃可见光透射比、太阳直接透射比、太阳能总透射比、紫外线透射比及有关窗玻璃参数测定》中的方法进行测定。其中初始透光率差值为初始褪色态透过率与初始着色态透过率的差值,循环20000次后的透光率差值为循环20000次后的褪色态透过率与循环20000次后的着色态透过率的差值。
[0118]
着色时间和褪色时间的测定方法为:将上述电致变色器件分别放入紫外-可见光谱仪中,在550nm处检测透过率最大值和透过率最小值,透过率由最大值变化为最小值所用的时间记为着色时间,透过率由最小值变化为最大值所用的时间记为褪色时间。
[0119]
表1
[0120]
[0121]
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:应用本发明的技术方案,具备上述式(i)或式(ii)所示结构的电致变色化合物组成的电致变色器件具备优异的循环稳定性,长时间多次着/褪色循环后性能无明显衰减,同时还具备变色速度快,不同状态下透光率差值高的特性,有效延长了使用寿命。
[0122]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1