催化聚合的双次膦酰胺金属配合物催化剂及其制备方法与流程
1.本发明涉及配合物催化剂技术领域,具体涉及催化聚合的双次膦酰胺金属配合物催化剂及其制备方法。
背景技术:
2.聚双环戊二烯由双环戊二烯在催化剂的作用下开环易位聚合而成,工业生产中常用的催化剂是以金属钨、钼类化合物的主催化剂以及助催化剂有机铝化合物组成的双组分催化体系,该类催化体系可配置成双物料反应体系,适合反应注射成型工艺,但是该类催化活性较低,并且对环境较为敏感从而容易失活。
3.公告号cn111732681b的发明专利公开了一种双组分潜伏型金属卡宾催化体系,由金属卡宾和三烃基膦的混合物与卤代烃组成;金属卡宾为钌金属卡宾、钼金属卡宾中的一种或多种;该双组分潜伏型金属卡宾催化体系在反应注射成型工艺的双物料反应体系中应用时,可延长双物料体系的存放时间,适合工业化催化合成pdcpd。但是经研究发现,存在以下技术问题:配体不能与过渡金属以多种配位模式得到稳定性好的金属配合物催化剂,制备方法不易于工业化应用。
技术实现要素:
4.本发明的目的在于提供一种催化聚合的双次膦酰胺金属配合物催化剂及其制备方法,用于解决现有技术中配体不能与过渡金属以多种配位模式得到稳定性好的金属配合物催化剂,制备方法不易于工业化应用的技术问题。
5.本发明的目的可以通过以下技术方案实现:
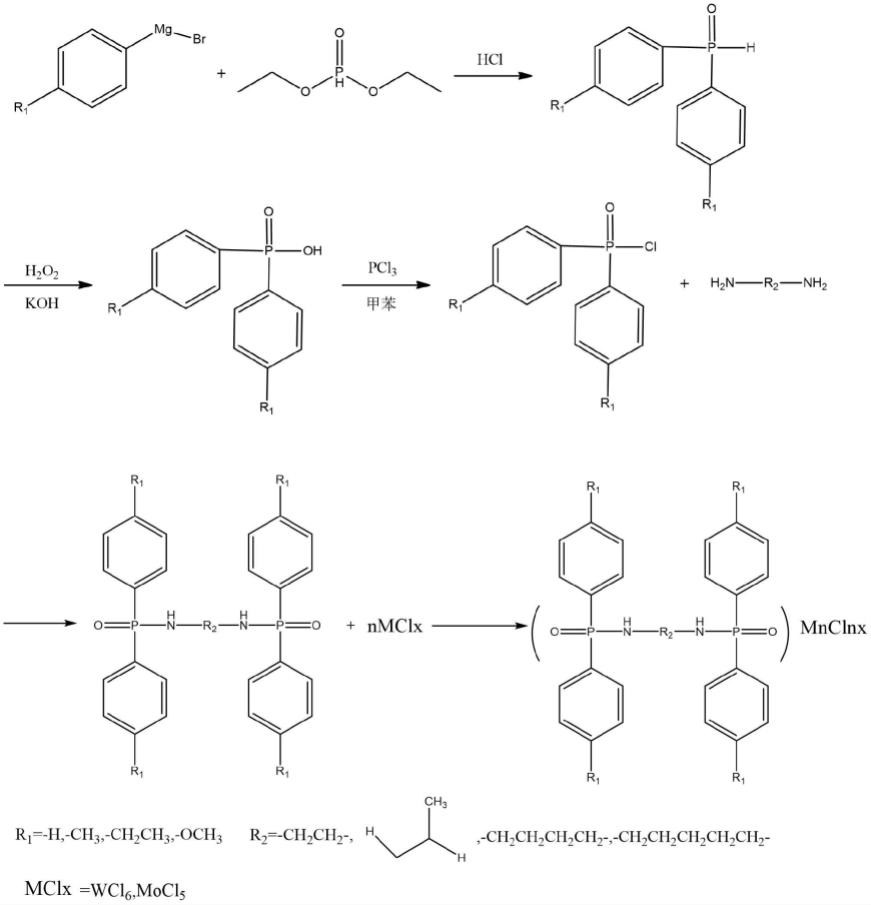
6.本发明提供一种催化聚合的双次膦酰胺金属配合物催化剂,以芳基溴化镁为原料,与亚磷酸二乙酯经芳基格式反应合成二芳基膦氧化物;二芳基膦氧化物在碱性条件下氧化反应合成二芳基次膦酸,二芳基次膦酸经氯代反应合成二芳基次膦酰氯,二芳基次膦酰氯与烷基二元胺合成双次膦酰胺配体,双次膦酰胺配体与金属卤化物合成得到该金属配合物催化剂。
7.本发明双次膦酰胺金属配合物催化剂的合成反应式如下:
[0008][0009]
进一步的,所述芳基溴化镁选自苯基溴化镁、对甲基苯基溴化镁、对乙基苯基溴化镁或对甲氧基苯基溴化镁;所述烷基二元胺选自乙二胺、1,2-丙二胺、1,4-丁二胺、1,5-戊二胺中的一种或多种的混合物;所述金属卤化物选自六氯化钨、五氯化钼中的一种或两种的混合物。
[0010]
本发明还提供了上述催化聚合的双次膦酰胺金属配合物催化剂的制备方法,包括以下步骤:
[0011]
步骤一,芳基格式反应:氮气保护下,向冰浴的三口烧瓶内加入浓度0.08~0.12mol/l的芳基溴化镁溶液,滴液漏斗滴加亚磷酸二乙酯的四氢呋喃溶液,搅拌10~20min后转移至72~80℃下油浴加热,回流反应1~2小时,静置至室温,氮气保护下加入25~40wt%的盐酸,直至反应液澄清后停止滴加;减压蒸馏除去四氢呋喃,加入乙酸乙酯萃取分层,无水硫酸镁干燥,过滤;滤液使用乙酸乙酯洗涤,减压蒸馏脱除乙酸乙酯,得到二芳基膦氧化物;
[0012]
步骤二,二芳基膦氧化物氧化反应:将二芳基膦氧化物、15~30wt%的氢氧化钾溶液加入配备回流冷凝管的三口烧瓶内,升温至90~98℃,恒压漏斗滴加30wt%的双氧水溶
液;滴加完毕后,升温至100~106℃,回流冷凝下保温搅拌1~1.5小时;反应液自然降温至室温,冰浴下加入25~40wt%的盐酸直至产生絮状物,水洗,乙酸乙酯洗,过滤,滤饼干燥得到二芳基次膦酸;
[0013]
步骤三,二芳基次膦酸氯代反应:将二芳基次膦酸和三氯化磷、甲苯依次加入三口烧瓶内,升温至75~85℃,保温搅拌反应3~5小时,减压浓缩除去甲苯,55~65℃干燥12~16小时,-5~10℃下使用乙酸乙酯与石油醚的混合溶剂重结晶得到二芳基次膦酰氯;
[0014]
步骤四,双次膦酰胺配体合成:将二芳基次膦酰氯和溶剂加入三口烧瓶内,冰水浴下滴加烷基二元胺的四氢呋喃溶液,滴加完毕后,升温至40~80℃,保温搅拌反应3~4小时;反应液减压浓缩除去溶剂和四氢呋喃,水洗3~5次,滤饼65~80℃干燥得到双次膦酰胺配体;
[0015]
步骤五,金属配合物催化剂合成:氮气保护下向三口烧瓶内加入双次膦酰胺配体和甲苯,搅拌溶解双次膦酰胺配体后,加入金属卤化物,升温至35~45℃,保温搅拌2~4小时,加入甲苯得到浓度0.1~0.5mol/l的金属配合物催化剂。
[0016]
进一步的,所述步骤一中芳基溴化镁选自苯基溴化镁、对甲基苯基溴化镁、对乙基苯基溴化镁或对甲氧基苯基溴化镁,芳基溴化镁溶液的溶剂为四氢呋喃或乙醚,芳基溴化镁与亚磷酸二乙酯的摩尔比为2.2~2.5:1,亚磷酸二乙酯与四氢呋喃的质量比为1:3~6。
[0017]
进一步的,所述步骤二中氢氧化钾溶液的用量为二芳基膦氧化物质量的1.5~4倍,双氧水溶液的用量为二芳基膦氧化物质量的0.6~2.5倍。
[0018]
进一步的,所述步骤三中二芳基次膦酸和三氯化磷的摩尔比为1:1.1~1.3,甲苯的用量为二芳基次膦酸质量的3~6倍;混合溶剂中乙酸乙酯与石油醚的体积比为1:2~3。
[0019]
进一步的,所述步骤四中二芳基次膦酰氯与烷基二元胺的摩尔比为2.3~2.6:1,溶剂为乙酰基二甲胺、甲苯、二甲苯、甲醇、二氯甲烷、乙腈、四氢呋喃中的一种或多种的混合物;烷基二元胺选自乙二胺、1,2-丙二胺、1,4-丁二胺、1,5-戊二胺中的一种或多种的混合物。
[0020]
进一步的,所述步骤五中金属卤化物选自六氯化钨、五氯化钼中的一种或两种的混合物。
[0021]
本发明具备下述有益效果:
[0022]
1、本发明的双次膦酰胺金属配合物催化剂,以芳基溴化镁为原料,与亚磷酸二乙酯经芳基格式反应、氧化反应、氯代反应合成二芳基次膦酰氯,二芳基次膦酰氯与烷基二元胺合成双次膦酰胺配体,双次膦酰胺配体与金属卤化物合成得到催化剂成品;由于双次膦酰胺配体中含有双n-p配位中心,能够与过渡金属以多种配位模式反应生成结构新颖的金属络合物,使得到的金属配合物催化剂稳定性好,高效催化双环戊二烯聚合得到性能稳定耐久的聚双环戊二烯制品。
[0023]
2、本发明双次膦酰胺金属配合物催化剂的制备方法,步骤二采用氢氧化钾、双氧水的碱性氧化体系,高转化率的得到高纯度的二芳基次膦酸;步骤三采用三氯化磷作为氯代试剂,能够高选择性氯代得到二芳基次膦酰氯,重结晶工序即使没有完全去除也不影响产物的纯度;步骤四通过控制反应温度和溶剂用量,高收率的得到双次膦酰胺配体;该制备方法方便操作,易于工业化应用。
具体实施方式
[0024]
下面将结合实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0025]
实施例1
[0026]
本实施例催化聚合的双次膦酰胺金属配合物催化剂的制备方法,包括以下步骤:
[0027]
步骤一,芳基格式反应:氮气保护下,向冰浴的三口烧瓶内加入浓度0.096mol/l的苯基溴化镁溶液2l,滴液漏斗滴加亚磷酸二乙酯的四氢呋喃溶液60.5g,亚磷酸二乙酯的用量为11g,搅拌16min后转移至78℃下油浴加热,回流反应1.5小时,静置至室温,氮气保护下加入32wt%的盐酸,直至反应液澄清后停止滴加;减压蒸馏除去四氢呋喃,加入乙酸乙酯萃取分层,无水硫酸镁干燥,过滤;滤液使用乙酸乙酯洗涤,减压蒸馏脱除乙酸乙酯,得到二芳基膦氧化物;其中,苯基溴化镁溶液的溶剂为四氢呋喃;
[0028]
步骤二,二芳基膦氧化物氧化反应:将二芳基膦氧化物25g、25wt%的氢氧化钾溶液71.3g加入配备回流冷凝管的三口烧瓶内,升温至95℃,恒压漏斗滴加30wt%的双氧水溶液19.5g;滴加完毕后,升温至103℃,回流冷凝下保温搅拌1.2小时;反应液自然降温至室温,冰浴下加入30wt%的盐酸直至产生絮状物,水洗,乙酸乙酯洗,过滤,滤饼干燥得到二芳基次膦酸;
[0029]
步骤三,二芳基次膦酸氯代反应:将二芳基次膦酸21.8g和三氯化磷16.5g、甲苯100.3g依次加入三口烧瓶内,升温至83℃,保温搅拌反应4.5小时,减压浓缩除去甲苯,62℃干燥15小时,-2℃下使用乙酸乙酯与石油醚的混合溶剂重结晶得到二芳基次膦酰氯;其中,混合溶剂中乙酸乙酯与石油醚的体积比为1:2.5;
[0030]
步骤四,双次膦酰胺配体合成:将二芳基次膦酰氯23.7g和溶剂甲苯80g加入三口烧瓶内,冰水浴下滴加1,4-丁二胺的四氢呋喃溶液18.5g,1,4-丁二胺的用量为3.5g,滴加完毕后,升温至55℃,保温搅拌反应3.5小时;反应液减压浓缩除去溶剂甲苯和四氢呋喃,水洗4次,滤饼72℃干燥得到双次膦酰胺配体;
[0031]
步骤五,金属配合物催化剂合成:氮气保护下向三口烧瓶内加入双次膦酰胺配体24.4g和甲苯60g,搅拌溶解双次膦酰胺配体后,加入六氯化钨21.8g,升温至40℃,保温搅拌2.5小时,加入甲苯得到浓度0.3mol/l的金属配合物催化剂。
[0032]
本实施例步骤四中双次膦酰胺配体的收率为76.5%。采用由70wt%双环戊二烯、20wt%苯乙烯、8.5wt%氧化石墨烯、0.8wt%抗氧化剂1010、0.7wt%紫外线吸收剂uv329组成的混合液,与本实施例制备的金属配合物催化剂,在70℃下注射成型,固化3min后得到聚双环戊二烯制品,常温放置2个月后粘度增加2.8%,金属配合物催化剂与双环戊二烯的摩尔比为250:1。
[0033]
实施例2
[0034]
本实施例催化聚合的双次膦酰胺金属配合物催化剂的制备方法,包括以下步骤:
[0035]
步骤一,芳基格式反应:氮气保护下,向冰浴的三口烧瓶内加入浓度0.086mol/l的对甲基苯基溴化镁溶液2l,滴液漏斗滴加亚磷酸二乙酯的四氢呋喃溶液51.6g,亚磷酸二乙酯的用量为10.3g,搅拌18min后转移至76℃下油浴加热,回流反应1.6小时,静置至室温,氮
气保护下加入32wt%的盐酸,直至反应液澄清后停止滴加;减压蒸馏除去四氢呋喃,加入乙酸乙酯萃取分层,无水硫酸镁干燥,过滤;滤液使用乙酸乙酯洗涤,减压蒸馏脱除乙酸乙酯,得到二芳基膦氧化物;其中,对甲基苯基溴化镁溶液的溶剂为乙醚;
[0036]
步骤二,二芳基膦氧化物氧化反应:将二芳基膦氧化物25g、22wt%的氢氧化钾溶液75g加入配备回流冷凝管的三口烧瓶内,升温至96℃,恒压漏斗滴加30wt%的双氧水溶液40g;滴加完毕后,升温至105℃,回流冷凝下保温搅拌1.2小时;反应液自然降温至室温,冰浴下加入36wt%的盐酸直至产生絮状物,水洗,乙酸乙酯洗,过滤,滤饼干燥得到二芳基次膦酸;
[0037]
步骤三,二芳基次膦酸氯代反应:将二芳基次膦酸24.6g和三氯化磷17.1g、甲苯123g依次加入三口烧瓶内,升温至82℃,保温搅拌反应4.5小时,减压浓缩除去甲苯,62℃干燥15.5小时,3℃下使用乙酸乙酯与石油醚的混合溶剂重结晶得到二芳基次膦酰氯;其中,混合溶剂中乙酸乙酯与石油醚的体积比为1:2.8;
[0038]
步骤四,双次膦酰胺配体合成:将二芳基次膦酰氯26.5g和溶剂二氯甲烷70g加入三口烧瓶内,冰水浴下滴加1,2-丙二胺的四氢呋喃溶液25g,1,2-丙二胺的用量为2.9g,滴加完毕后,升温至65℃,保温搅拌反应3.6小时;反应液减压浓缩除去溶剂和四氢呋喃,水洗5次,滤饼72℃干燥得到双次膦酰胺配体;
[0039]
步骤五,金属配合物催化剂合成:氮气保护下向三口烧瓶内加入双次膦酰胺配体26.5g和甲苯70g,搅拌溶解双次膦酰胺配体后,加入五氯化钼20.5g,升温至42℃,保温搅拌3.5小时,加入甲苯得到浓度0.4mol/l的金属配合物催化剂。
[0040]
本实施例步骤四中双次膦酰胺配体的收率为73.8%。采用由70wt%双环戊二烯、20wt%苯乙烯、8.5wt%氧化石墨烯、0.8wt%抗氧化剂1010、0.7wt%紫外线吸收剂uv329组成的混合液与本实施例制备的金属配合物催化剂,在70℃下注射成型,固化2min后得到聚双环戊二烯制品,常温放置2个月后粘度增加3.2%,金属配合物催化剂与双环戊二烯的摩尔比为230:1。
[0041]
实施例3
[0042]
本实施例催化聚合的双次膦酰胺金属配合物催化剂的制备方法,包括以下步骤:
[0043]
步骤一,芳基格式反应:氮气保护下,向冰浴的三口烧瓶内加入浓度0.11mol/l的对乙基苯基溴化镁溶液2l,滴液漏斗滴加亚磷酸二乙酯的四氢呋喃溶液53.3g,亚磷酸二乙酯的用量为12.7g,搅拌17min后转移至78℃下油浴加热,回流反应1.8小时,静置至室温,氮气保护下加入28wt%的盐酸,直至反应液澄清后停止滴加;减压蒸馏除去四氢呋喃,加入乙酸乙酯萃取分层,无水硫酸镁干燥,过滤;滤液使用乙酸乙酯洗涤,减压蒸馏脱除乙酸乙酯,得到二芳基膦氧化物;其中,对甲基苯基溴化镁溶液的溶剂为四氢呋喃;
[0044]
步骤二,二芳基膦氧化物氧化反应:将二芳基膦氧化物25g、24wt%的氢氧化钾溶液75g加入配备回流冷凝管的三口烧瓶内,升温至96℃,恒压漏斗滴加30wt%的双氧水溶液55g;滴加完毕后,升温至104℃,回流冷凝下保温搅拌1.5小时;反应液自然降温至室温,冰浴下加入32wt%的盐酸直至产生絮状物,水洗,乙酸乙酯洗,过滤,滤饼干燥得到二芳基次膦酸;
[0045]
步骤三,二芳基次膦酸氯代反应:将二芳基次膦酸27.4g和三氯化磷17.2g、甲苯142.4g依次加入三口烧瓶内,升温至82℃,保温搅拌反应4.5小时,减压浓缩除去甲苯,62℃
干燥14.5小时,6℃下使用乙酸乙酯与石油醚的混合溶剂重结晶得到二芳基次膦酰氯;其中,混合溶剂中乙酸乙酯与石油醚的体积比为1:2.8;
[0046]
步骤四,双次膦酰胺配体合成:将二芳基次膦酰氯29.3g和溶剂乙腈80g加入三口烧瓶内,冰水浴下滴加1,5-戊二胺的四氢呋喃溶液28g,1,5-戊二胺的用量为3.9g,滴加完毕后,升温至57℃,保温搅拌反应3.8小时;反应液减压浓缩除去溶剂和四氢呋喃,水洗4次,滤饼76℃干燥得到双次膦酰胺配体;
[0047]
步骤五,金属配合物催化剂合成:氮气保护下向三口烧瓶内加入双次膦酰胺配体30.7g和甲苯90g,搅拌溶解双次膦酰胺配体后,加入六氯化钨35.7g,升温至43℃,保温搅拌3.6小时,加入甲苯得到浓度0.42mol/l的金属配合物催化剂。
[0048]
本实施例步骤四中双次膦酰胺配体的收率为76.8%。采用由70wt%双环戊二烯、20wt%苯乙烯、8.5wt%氧化石墨烯、0.8wt%抗氧化剂1010、0.7wt%紫外线吸收剂uv329组成的混合液与本实施例制备的金属配合物催化剂,在65℃下注射成型,固化4min后得到聚双环戊二烯制品,常温放置2个月后粘度增加3.5%,金属配合物催化剂与双环戊二烯的摩尔比为220:1。
[0049]
实施例4
[0050]
本实施例催化聚合的双次膦酰胺金属配合物催化剂的制备方法,包括以下步骤:
[0051]
步骤一,芳基格式反应:氮气保护下,向冰浴的三口烧瓶内加入浓度0.12mol/l的对甲氧基苯基溴化镁溶液2l,滴液漏斗滴加亚磷酸二乙酯的四氢呋喃溶液81g,亚磷酸二乙酯的用量为13.5g,搅拌20min后转移至80℃下油浴加热,回流反应1.8小时,静置至室温,氮气保护下加入36wt%的盐酸,直至反应液澄清后停止滴加;减压蒸馏除去四氢呋喃,加入乙酸乙酯萃取分层,无水硫酸镁干燥,过滤;滤液使用乙酸乙酯洗涤,减压蒸馏脱除乙酸乙酯,得到二芳基膦氧化物;其中,对甲氧基苯基溴化镁溶液的溶剂为乙醚;
[0052]
步骤二,二芳基膦氧化物氧化反应:将二芳基膦氧化物25g、28wt%的氢氧化钾溶液90g加入配备回流冷凝管的三口烧瓶内,升温至97℃,恒压漏斗滴加30wt%的双氧水溶液52.5g;滴加完毕后,升温至106℃,回流冷凝下保温搅拌1.5小时;反应液自然降温至室温,冰浴下加入37wt%的盐酸直至产生絮状物,水洗,乙酸乙酯洗,过滤,滤饼干燥得到二芳基次膦酸;
[0053]
步骤三,二芳基次膦酸氯代反应:将二芳基次膦酸27.8g和三氯化磷17.6g、甲苯144.6g依次加入三口烧瓶内,升温至85℃,保温搅拌反应4.6小时,减压浓缩除去甲苯,64℃干燥15.5小时,8℃下使用乙酸乙酯与石油醚的混合溶剂重结晶得到二芳基次膦酰氯;其中,混合溶剂中乙酸乙酯与石油醚的体积比为1:3。
[0054]
步骤四,双次膦酰胺配体合成:将二芳基次膦酰氯29.7g和溶剂乙酰基二甲胺85g加入三口烧瓶内,冰水浴下滴加乙二胺的四氢呋喃溶液10g,乙二胺的用量为2.3g,滴加完毕后,升温至46℃,保温搅拌反应4小时;反应液减压浓缩除去溶剂和四氢呋喃,水洗5次,滤饼76℃干燥得到双次膦酰胺配体;
[0055]
步骤五,金属配合物催化剂合成:氮气保护下向三口烧瓶内加入双次膦酰胺配体29g和甲苯110g,搅拌溶解双次膦酰胺配体后,加入五氯化钼20.5g,升温至44℃,保温搅拌3.8小时,加入甲苯得到浓度0.38mol/l的金属配合物催化剂。
[0056]
本实施例步骤四中双次膦酰胺配体的收率为73.7%。采用由70wt%双环戊二烯、
20wt%苯乙烯、8.5wt%氧化石墨烯、0.8wt%抗氧化剂1010、0.7wt%紫外线吸收剂uv329组成的混合液与本实施例制备的金属配合物催化剂,在75℃下注射成型,固化2min后得到聚双环戊二烯制品,常温放置2个月后粘度增加2.8%,金属配合物催化剂与双环戊二烯的摩尔比为210:1。
[0057]
对比例1
[0058]
本对比例与实施例4的区别在于,步骤二中氢氧化钾溶液替换为碳酸钾溶液。
[0059]
本对比例采用由70wt%双环戊二烯、20wt%苯乙烯、8.5wt%氧化石墨烯、0.8wt%抗氧化剂1010、0.7wt%紫外线吸收剂uv329组成的混合液,与制备的金属配合物催化剂在70℃下注射成型,固化1min后得到聚双环戊二烯制品,常温放置2个月后粘度增加5.3%,金属配合物催化剂与双环戊二烯的摩尔比为210:1。
[0060]
对比例2
[0061]
本对比例与实施例4的区别在于,步骤三中重结晶溶剂选自乙醇与石油醚按照体积比1:3组成的混合溶剂。
[0062]
本对比例采用由70wt%双环戊二烯、20wt%苯乙烯、8.5wt%氧化石墨烯、0.8wt%抗氧化剂1010、0.7wt%紫外线吸收剂uv329组成的混合液,与制备的金属配合物催化剂在70℃下注射成型,固化1min后得到聚双环戊二烯制品,常温放置2个月后粘度增加5.8%,金属配合物催化剂与双环戊二烯的摩尔比为210:1。
[0063]
对比例3
[0064]
本对比例与实施例4的区别在于,步骤五中加入四氢呋喃得到浓度0.38mol/l的金属配合物催化剂。
[0065]
本对比例采用由70wt%双环戊二烯、20wt%苯乙烯、8.5wt%氧化石墨烯、0.8wt%抗氧化剂1010、0.7wt%紫外线吸收剂uv329组成的混合液,与制备的金属配合物催化剂在70℃下注射成型,固化1min后得到聚双环戊二烯制品,常温放置2个月后粘度增加5.6%,金属配合物催化剂与双环戊二烯的摩尔比为210:1。
[0066]
对比例1由于步骤二中氢氧化钾溶液替换为碳酸钾溶液,无法高转化率的得到高纯度的二芳基次膦酸;对比例2由于更换重结晶溶剂,无法得到高纯度的二芳基次膦酰氯;对比例3由于加入四氢呋喃得到金属配合物催化剂,使得金属配合物催化剂的溶剂成分改变。对比例1-3的金属配合物催化剂在催化双环戊二烯聚合生成聚双环戊二烯时,常温放置后粘度增加较大,性能耐久性降低。
[0067]
以上公开的本发明优选实施例只是用于帮助阐述本发明。优选实施例并没有详尽叙述所有的细节,也不限制该发明仅为的具体实施方式。显然,根据本说明书的内容,可做很多的修改和变化。本说明书选取并具体描述这些实施例,是为了更好地解释本发明的原理和实际应用,从而使所属技术领域技术人员能很好地理解和利用本发明。本发明仅受权利要求书及其全部范围和等效物的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1