FGFR抑制剂及其制备方法、药物组合物和应用
本发明涉及一种fgfr抑制剂及其制备方法、药物组合物和应用,尤其涉及一种对耐药突变体fgfrv550l具有优异的特异性抑制活性的fgfr抑制剂及其制备方法、药物组合物和应用。
背景技术:
1、成纤维细胞生长因子受体(fgfr)属于受体酪氨酸激酶(rtks)家族。通过与其配体结合,即成纤维细胞生长因子(fgf),fgfr可以在细胞膜上形成一个同源二聚体。这些二聚体可引起fgfr胞内关键酪氨酸残基的磷酸化,从而激活细胞内的一些下游信号传导通路。这些细胞内信号通路在细胞增殖、生存和分化中发挥着重要作用。fgfr信号转导途径的紊乱,包括配体和受体的表达增加、fgfr基因扩增和突变、缺失等改变,可以促进细胞的致癌转化,在肿瘤细胞增殖、耐药和血管生成中发挥重要作用。例如,在30%的肝癌中发现了fgfr4基因的扩增;在10%-20%的胆管癌发现了fgfr2基因的融合;在10%-60%的尿路上皮癌中发现了fgfr3基因的突变;在10%的非小细胞肺癌(nsclc)中发现了fgfr1基因的扩增等改变等。由于fgfr在肿瘤的发生和发展过程中起着关键性的作用,开发新的具有高抑制活性和优异药代动力学性质的fgfr激酶抑制剂已成为开发新型抗肿瘤药物的关键。
2、截至目前,仅有三款靶向fgfr的小分子抑制剂(erdafitinib、infigratinib和pemigatinib)上市,多款小分子抑制剂临床在研(如futibatinib、fisogatinib等)。临床试验表明,fgfr4选择性抑制剂fisogatinib(blu-554)对fgf19过表达的患者显示出了临床效益和肿瘤发生消退。pemigatinib被美国fda批准用于既往接受过治疗的携带fgfr2融合/重排的局部晚期或转移性胆管癌患者。infigratinib被美国fda批准用于既往接受过治疗且携带fgfr2基因融合或其它重排类型的不可切除局部晚期或转移性胆管癌成人患者。
3、尽管fgfr是经验证的治疗的癌症药物靶点和生物标志物,但是在上述药物在临床应用中存在一定比例的耐药现象。患者在使用fisogatinib抑制剂治疗6个月后,观察到有29%的患者对这些小分子抑制剂的抗性。fgfr4耐药突变的频率与非小细胞肺癌中的egfr和alk突变的频率相当。其中最突出的耐药突变是门卫残基v550l和v550m。这种获得性突变极大降低了药物与靶点的亲和力,从而产生耐药性,并导致肿瘤复发或疾病进展。同样pemigatinib与infigratinib都存在门卫残基突变,产生获得性耐药的问题。
技术实现思路
1、发明目的:针对现有化合物对门卫残基发生突变的fgfr亚型抑制活性不足等问题,本发明旨在提供一种针对耐药突变体fgfrv550l具有显著特异性抑制作用的fgfr抑制剂及其制备方法、药物组合物和应用。
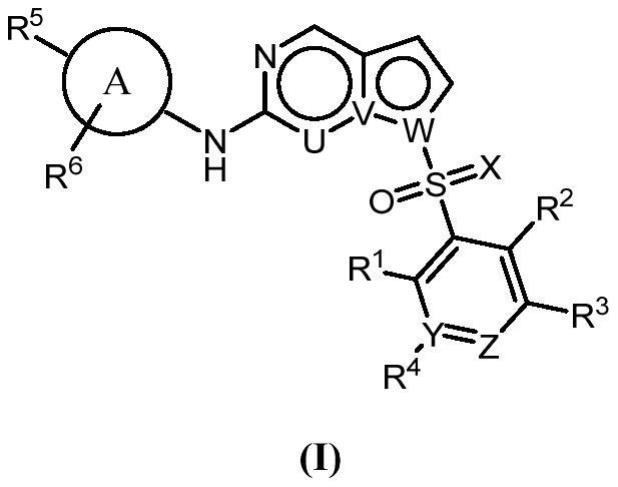
2、技术方案:作为本发明涉及的第一方面,本发明的fgfr抑制剂具有式i的结构,所述fgfr抑制剂包含其立体异构体、药学上可接受的盐或它们的混合物:
3、
4、其中,u选自ch或n;
5、v、w各自独立地选自c或n且不相同;
6、x选自o、nh、nch3、ncn或ncf3;
7、y选自n或c;
8、z选自n或ch;
9、a环选自芳基或芳杂基;所述芳基选自苯基、萘基、苊基或四氢萘基;所述芳杂基选自哌啶基、吡咯基、吡唑基、咪唑基、呋喃基、噻吩基、噁唑基、异噁唑基、噻唑基、异噻唑基、吡啶基、嘧啶基、吡嗪基、哒嗪基、喹啉基、喹唑啉基、吲哚基、苯并咪唑基、苯并噁唑基、苯并异噁唑基、苯并噻唑基、苯并异噻唑基、苯并呋喃基、苯并噻吩基、2,3-二氢苯并[1,4]二氧杂环己烯基或苯并[1,3]二氧杂环戊烯基;
10、r1、r2、r3、r4、r6各自独立地选自h、卤素、ch2oh、cf3、ocf3、cn、c1-6烷基或c1-6烷氧基;或者r2、r3连接在一起形成3-8元杂环,所述3-8元杂环可任选地被c1-6烷基取代;
11、r5选自-ora、-(ch2)mora、-nrarb、-conrarb、-so2nrarb、-(sonh)nrarb、-(ch2)m-nrarb、-(ch2)mcon(ch3)2、-(ch2)mso2n(ch3)2或3-10元杂环,所述3-10元杂环可任选地被1-3个r7取代;m=1、2或3;
12、ra、rb各自独立地选自h、c1-6烷基、-coc1-5烷基或-so2c1-5烷基,所述c1-6烷基、-coc1-5烷基、-so2c1-5烷基任选地被-nrcrd、-conrcrd或-so2c1-5烷基取代;
13、rc、rd各自独立地选自h、c1-6烷基或-coc1-5烷基;
14、r7选自c1-6烷基、c3-8环烷基、-conh2、-so2nh2、-ch2cooh、-ch2so3h、-ch2cn、-(ch2)2nrcrd、-nrcrd、-(ch2)noh、-cooc1-6烷基或3-8元杂环,所述3-8元杂环可任选地被c1-6烷基取代;n=1、2或3。
15、进一步地,上述fgfr抑制剂具有式ia或ib的结构:
16、
17、其中a、x、y、z、r1~r6的定义如权利要求1所述。
18、优选,上述结构中:
19、x选自o或nh;
20、y选自n或c;
21、z选自n或ch;
22、a环选自芳基或芳杂基;所述芳基选自苯基、萘基、苊基或四氢萘基;所述芳杂基选自哌啶基、吡咯基、吡唑基、咪唑基、呋喃基、噻吩基、噁唑基、异噁唑基、噻唑基、异噻唑基、吡啶基、嘧啶基、吡嗪基、哒嗪基、喹啉基、喹唑啉基、吲哚基、苯并咪唑基、苯并吡唑基、苯并异噁唑基、苯并噻唑基、苯并异噻唑基、苯并呋喃基、苯并噻吩基、2,3-二氢苯并[1,4]二氧杂环己烯基或苯并[1,3]二氧杂环戊烯基;
23、r1、r2各自独立地选自h、卤素、cn或c1-6烷氧基;
24、r3、r4各自独立地选自h、卤素、ocf3或c1-6烷氧基;或者r2、r3连接在一起形成5-6元单环烷基或5-6元单环芳杂基;所述5-6元单环烷基选自四氢呋喃基、吡咯烷基、1,3-二噁茂基、1,3-噁唑烷基或哌啶基;所述5-6元单环芳杂基选自哌啶基、吡咯基、吡唑基、咪唑基、呋喃基、噻吩基、噁唑基、异噁唑基、噻唑基、异噻唑基、吡啶基、嘧啶基、吡嗪基或哒嗪基;
25、r6选自h、卤素、ch2oh、c1-6烷基或c1-6烷氧基;
26、r5选自-ora、-(ch2)mora、-nrarb、-conrarb、-so2nrarb、-(sonh)nrarb、-(ch2)m-nrarb、-(ch2)mcon(ch3)2、-(ch2)mso2n(ch3)2或3-10元杂环,所述3-10元杂环可任选地被1-3个r7取代;m=1、2或3;
27、ra、rb各自独立地选自h、c1-6烷基、-coc1-5烷基或-so2c1-5烷基,所述c1-6烷基、-coc1-5烷基、-so2c1-5烷基任选地被-nrcrd、-conrcrd或-so2c1-5烷基取代;
28、rc、rd各自独立地选自h、c1-6烷基或-coc1-5烷基;
29、r7选自c1-6烷基、c3-8环烷基、-conh2、-so2nh2、-ch2cooh、-ch2so3h、-ch2cn、-(ch2)2nrcrd、-nrcrd、-(ch2)noh、-cooc1-6烷基或3-8元杂环,所述3-8元杂环任选地被c1-6烷基取代;n=1、2或3。
30、进一步优选,上述结构中:
31、x选自o;
32、y选自n或c;
33、z选自n或ch;
34、a环选自苯基、吡唑基、咪唑基、吡啶基、嘧啶基、苯并咪唑基、苯并吡唑基或苯并异噁唑基;
35、r1、r2各自独立地选自h、f、cl、cn或c1-3烷氧基;
36、r3、r4各自独立地选自h、f、cl或c1-3烷氧基;或者r2、r3连接在一起形成5-6元单环烷基或5-6元单环芳杂基;所述5-6元单环烷基选自1,3-二噁茂基、1,3-噁唑烷基或哌啶基;所述5-6元单环芳杂基选自吡唑基、咪唑基、异噁唑基或噻唑基;
37、r6选自h、f、cl或c1-3烷氧基;
38、r5选自-ora、-(ch2)mora、-nrarb、-conrarb、-so2nrarb、-(sonh)nrarb、-(ch2)m-nrarb、-(ch2)mcon(ch3)2、-(ch2)mso2n(ch3)2或3-10元杂环,所述3-10元杂环可任选地被1-3个r7取代;m=1、2或3;
39、ra、rb各自独立地选自h、c1-6烷基、-coc1-5烷基或-so2c1-5烷基,所述c1-6烷基、-coc1-5烷基、-so2c1-5烷基任选地被-nrcrd、-conrcrd或-so2c1-5烷基取代;
40、rc、rd各自独立地选自h、c1-6烷基或-coc1-5;
41、r7选自c1-6烷基、c3-8环烷基、-conh2、-so2nh2、-ch2cooh、-ch2so3h、-ch2cn、-(ch2)2nrcrd、-nrcrd、-(ch2)noh、-cooc1-6烷基或3-8元杂环,所述3-8元杂环任选地被c1-6烷基取代;n=1、2或3。
42、更进一步优选,上述结构中:
43、x选自o;
44、y选自n或c;
45、z选自n或ch;
46、a环选自苯基、吡唑基、咪唑基、吡啶基、嘧啶基、苯并咪唑基、苯并吡唑基或苯并异噁唑基;
47、r1、r2各自独立地选自h、f、cl、cn或c1-3烷氧基;
48、r3、r4各自独立地选自h、f、cl或c1-3烷氧基;或者r2、r3连接在一起形成5-6元单环烷基或5-6元单环芳杂基;所述5-6元单环烷基选自1,3-二噁茂基、1,3-噁唑烷基或哌啶基;所述5-6元单环芳杂基选自吡唑基、咪唑基、异噁唑基或噻唑基;
49、r6选自h、f、cl或c1-3烷氧基;
50、r5选自
51、最优选,上述的fgfr抑制剂选自下列任一化合物:
52、
53、
54、
55、
56、
57、
58、
59、上述fgfr抑制剂药学上可接受的盐包括通式i化合物与下列酸形成的盐:盐酸、氢溴酸、硫酸、磷酸、甲磺酸、苯磺酸、对甲苯磺酸、萘磺酸、拧檬酸、酒石酸、乳酸、丙酮酸、乙酸、马来酸或琥珀酸、富马酸、水杨酸、苯基乙酸、杏仁酸;还包括通式i化合物与下列碱形成的盐:碱性金属阳离子盐、碱土金属阳离子盐或铵阳离子盐。
60、作为本发明涉及的第二方面,上述fgfr抑制剂的制备方法选自下列任一方法:
61、(1)当u、w为n,v为c时,以溴代苯或溴代吡啶为起始原料,经过取代、卤代、酰化反应制备而成通式化合物ia,
62、
63、(2)当u、w为n,v为c时,以溴代苯或溴代吡啶为起始原料,经过取代、卤代、酰化、取代反应制备而成通式化合物ia,
64、
65、(3)当u、v为n,w为c时,卤代吡咯并[2,1-f][1,2,4]三嗪为起始原料,经过两步取代、氧化、反应制备而成通式化合物ib,
66、
67、其中,y、z、r1~r6的定义如前所述;
68、将上述方法制备而成的化合物ia或ib与相应的酸或碱成盐,即得其药学上可接受的盐。
69、作为本发明涉及的第三方面,上述fgfr抑制剂与药学上可接受的载体形成药物组合物,具体的制剂形式如片剂、胶囊、糖浆、悬浮剂或注射剂,制剂可以加入香料、甜味剂、液体/固体填料、稀释剂等常用药用辅料。
70、作为本发明涉及的第四方面,上述fgfr抑制剂及其药物组合物应用于制备预防和/或治疗fgfr相关疾病的药物,具体为预防和/或治疗肝癌、非小细胞肺癌、胃癌、膀胱癌、头颈癌、胆管癌、乳腺癌、子宫内膜癌、宫颈癌、食道癌、肾癌、急性白血病、前列腺癌、甲状腺癌、皮肤癌、结肠直肠癌、胰腺癌、卵巢癌、骨髓增生异常综合症或间皮瘤的药物。
71、有益效果:与现有技术相比,本发明具有如下显著优点:
72、(1)该类fgfr抑制剂及其药物组合物可有效抑制野生型fgfr、突变型fgfr4v550l和huh7细胞增殖,ic50值最优均低于50nm;
73、(2)该类fgfr抑制剂及其药物组合物具有优异的肝微粒体稳定性,体内半衰期长、清除率低,生物利用度高,auc合适,具有较好的类药性质;
74、(3)该类fgfr抑制剂及其药物组合物应用广泛,可制备为治疗和/或预防与fgfr相关疾病的药物,尤其适用于对常规fgfr抑制剂耐药的各种肿瘤;所述药物在分子水平和细胞水平均可以发挥药效,并且治疗效果更优异,最优可达到十纳摩尔浓度级别水平;
75、(4)化合物制备方法简便、易操作。
- 还没有人留言评论。精彩留言会获得点赞!