一种胞内构建的CTSB响应的T2型小分子磁共振造影剂及其制备方法
一种胞内构建的ctsb响应的t2型小分子磁共振造影剂及其制备方法
技术领域
1.本发明属于磁共振成像技术领域,具体涉及可在高表达组织蛋白酶b(ctsb)的癌细胞内发生2-氰基苯并噻唑与d型半胱氨酸间点击缩合反应从而原位形成基于钆的纳米结构的小分子t2型磁共振造影剂的制备方法。
背景技术:
2.据《德国应用化学》杂志(angew.chem.int.ed.2017,56,15633-15638)报道组织蛋白酶b(ctsb)已被发现在各种恶性肿瘤中广泛过表达,如黑素瘤、乳腺癌、结肠癌、胃癌、头颈癌、肝癌、肺癌、卵巢癌、胰腺癌和前列腺癌。《德国应用化学》杂志(angew.chem.int.ed.2014,53(38),10077-10081)还报道发现ctsb在许多癌症类型的早期阶段表达增加,因此它已成为一种有吸引力的生物标志物,用于癌症的早期诊断。到目前为止,已经开发了许多成像技术来检测或成像ctsb。《分析化学》杂志报道了利用生物发光成像技术(anal.chem.2019,91,14834-14837)或荧光/光声成像技术(anal.chem.2021,93(27),9304-9308)来检测ctsb的活性;《德国应用化学》杂志(angew.chem.int.ed.2017,56(49),15633-15638)报道了利用化学发光成像技术来检测ctsb的活性。虽然这些光学成像技术具有操作简单、灵敏度高的特点,但是由于组织自身荧光和组织穿透深度较浅,因此这些光学成像技术特异性较差,不适用于组织深部肿瘤的诊断。相较于上述的光学成像技术,磁共振成像(magnetic resonance imaging,mri)作为一种先进的无创成像技术,可以提供三维详细的解剖图像,具有无限的组织穿透深度和极好的时空分辨率。《分析化学》杂志报道了临床上常用的磁共振造影剂一般为顺磁性钆基螯合物,如gd(iii)-二亚乙基三胺五乙酸(gd-dtpa)和gd(iii)-四氮杂环十二烷四乙酸(gd-dota)(anal.chem.2017,89,6922-6925),这些小分子反应物虽然可以很容易地穿过细胞膜,被细胞摄取,但是很快会被清除掉,从而影响其在细胞中的滞留时间。相对于小分子反应物,纳米结构在细胞中的保留时间长,但其存在很难被细胞摄取的问题。《自然
·
化学》(nat.chem.2010,2,54-60)报道的2-氰基苯并噻唑(cbt)与d型半胱氨酸(cys)间的点击缩合反应,通过该反应,小分子进入细胞后可转化为两亲性的寡聚体,进一步自组装成纳米结构,很难被细胞泵出。这种利用小分子前体在细胞内组装成纳米结构的智能策略,结合了小分子和纳米结构互补的优势,在分子成像和药物传递方面显示出极大的优势。另外,该点击缩合反应所触发的自组装过程具有条件温和、反应速度快且生物兼容性好的优点,而且可以通过控制ph、酶、以及还原型谷胱甘肽(gsh)等多种生物信号,从而灵活广泛地用于蛋白质标记、纳米材料的制备,分子成像和癌症治疗等方面。本发明课题组同学在《德国应用化学》杂志(angew.chem.int.ed.2022,https://doi.org/10.1002/anie.202114766)上报道了一种近红外光声探针,它利用2-氰基苯并噻唑与d型半胱氨酸间的点击缩合反应在细胞内形成纳米结构,增强了肿瘤的光声成像,从而用来检测ctsb的活性,但由于其探针分子水溶性较差以及在还原条件下不稳定,限制了它的发展和推广。为了解决这一问题,在此基础上,本发明通过将上述近红外光声探
针分子进行改进,获得了具有良好水溶性和稳定性的探针分子,即本发明的胞内构建的ctsb响应的t2型小分子磁共振造影剂。且至今尚未见到利用2-氰基苯并噻唑与d型半胱氨酸间点击缩合反应原位形成基于钆(gd)的纳米结构用于增强t2加权的磁共振肿瘤成像从而来诊断ctsb相关癌症方面研究工作的文献报道。
技术实现要素:
3.本发明的目的是提出一种可在高表达ctsb的癌细胞内发生2-氰基苯并噻唑与d型半胱氨酸间点击缩合反应从而原位形成基于钆的纳米结构的小分子t2型磁共振造影剂及其制备方法,以用于增强t2加权的磁共振肿瘤成像,填补利用点击缩合反应原位形成基于钆的纳米结构以增强t2加权的磁共振肿瘤成像从而来诊断ctsb相关癌症方面的空白。
4.本发明的胞内构建的ctsb响应的t2型小分子磁共振造影剂,其特征在于含有ctsb特异性酶切底物(缬氨酸-瓜氨酸,val-cit)、具有双硫键的潜在半胱氨酸(cys)基序和2-氰基-苯并噻唑(cbt)结构以及与赖氨酸(lys)侧链结合的1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸钆(dota-gd)结构的小分子化合物,分子式为val-cit-cys(set)-lys(dota-gd)-cbt,简记为vc-gd-cbt。
5.本发明的胞内构建的ctsb响应的t2型小分子磁共振造影剂的制备方法,其特征在于包括如下步骤:
6.第一步:将2mmol(937mg)的氨基酸boc-lys(fmoc)-oh和4mmol(443μl)的4-甲基吗啉(mmp)溶于4ml的无水四氢呋喃(thf)中,冷却至0℃,加入2mmol(254μl)的氯甲酸异丁酯(ibcf),在0℃的条件下反应混合物搅拌一个小时;然后将1.67mmol的2-氰基-6-氨基苯并噻唑(cbt)溶液(溶剂为无水thf)加入到反应混合物中,先在0℃下进一步搅拌一个小时,然后在室温下继续搅拌过夜;经过高效液相色谱分离纯化,得到第一种纯化合物,命名为化合物a;
7.第二步:将1.36mmol(848.5mg)的化合物a溶解在含有4ml二氯甲烷(dcm)、76ml三氟乙酸(tfa)和800μl三异丙基硅烷(tips)的混合溶液中(其中三氟乙酸和二氯甲烷的体积比为95:5,三异丙基硅烷的体积为溶液总体积的百分之一),室温下搅拌三个小时去除化合物a的叔丁氧羰基(boc)保护基团;经过高效液相色谱分离纯化,得到第二种纯化合物,命名为化合物b;
8.第三步:将1.17mmol(433.7mg)的氨基酸boc-cys(set)-oh、1.17mmol(158.3mg)的1-羟基苯并三唑(hobt)、1.17mmol(444.3mg)的o-苯并三氮唑-四甲基脲六氟磷酸酯(hbtu)、1.17mmol(204μl)的n,n-二异丙基乙胺(dipea)和0.78mmol(410.2mg)的化合物b溶解在n,n-二甲基甲酰胺中,室温下搅拌12个小时;经过高效液相色谱分离纯化,得到第三种纯化合物,命名为化合物c;
9.第四步:将0.54mmol(425.7mg)的化合物c溶解在含有2.5ml二氯甲烷(dcm)、47.5ml三氟乙酸(tfa)和500μl三异丙基硅烷(tips)的混合溶液中,室温下搅拌三个小时去除化合物c的叔丁氧羰基(boc)保护基团;经过高效液相色谱分离纯化,得到第四种纯化合物,命名为化合物d;
10.第五步:将0.62mmol(231.8mg)的氨基酸boc-val-cit-oh、0.87mmol(118mg)的1-羟基苯并三唑(hobt)、0.87mmol(330mg)的o-苯并三氮唑-四甲基脲六氟磷酸酯(hbtu)、
0.87mmol(152μl)的n,n-二异丙基乙胺(dipea)和0.58mmol(395.9mg)的化合物d溶解在n,n-二甲基甲酰胺中,室温下搅拌24个小时;经过高效液相色谱分离纯化,得到第五种纯化合物,命名为化合物e;
11.第六步:将化合物e的n-芴甲氧羰酰基(fmoc)保护基团用含10%哌啶的n,n-二甲基甲酰胺溶液(0.6ml哌啶+5.4ml的n,n-二甲基甲酰胺)在0℃下搅拌反应10分钟,然后加入540μl三氟乙酸用于中和其中的碱;经过高效液相色谱分离纯化,得到第六种纯化合物,命名为化合物f;
12.第七步:将0.38mmol(217.8mg)的三叔丁基1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸(dota(otbu)3)、0.38mmol(51.3mg)的1-羟基苯并三唑(hobt)和0.38mmol(144.1mg)的o-苯并三氮唑-四甲基脲六氟磷酸酯(hbtu)溶解于2毫升的n,n-二甲基甲酰胺中,随后加入0.38mmol(66μl)的n,n-二异丙基乙胺(dipea),室温下搅拌0.5个小时,然后将溶解于n,n-二甲基甲酰胺中的0.32mmol(260.6mg)的化合物f加入上述溶液中,室温下继续搅拌过夜;经过高效液相色谱分离纯化,得到第七种纯化合物,命名为化合物g;
13.第八步:将0.19mmol(263.5mg)的化合物g溶解在含有1.5ml二氯甲烷(dcm)、28.5ml三氟乙酸(tfa)和300μl三异丙基硅烷(tips)的混合溶液中,室温下搅拌三个小时去除化合物g的叔丁氧羰基(boc)和氧叔丁基(otbu)保护基团;经过高效液相色谱分离纯化,得到第八种纯化合物,命名为化合物h;
14.第九步:将0.043mmol(15.9mg)的氯化钆(gdcl3·
6h2o)溶解在水中,并用饱和碳酸钠溶液调节ph值为6.5-7.2,然后将其加入到0.02mmol(22.2mg)化合物h的n,n-二甲基甲酰胺溶液中,并将该混合溶液的ph值调节至6.5-7.2,在室温下搅拌反应3个小时。经过高效液相色谱分离纯化,即可得到基于gd的小分子t2型造影剂vc-gd-cbt。
15.在本发明的上述方法中合成的化合物a、b、c、d、e和f,其结构分别为:
[0016][0017][0018]
在本发明的上述方法中合成的化合物g、h和基于gd的小分子t2型造影剂vc-gd-cbt,其特征结构分别为:
[0019][0020]
与临床用的小分子造影剂相比,本发明的胞内构建的ctsb响应的t2型小分子磁共振造影剂,含有ctsb特异性酶切底物、具有双硫键的潜在半胱氨酸基序和2-氰基-苯并噻唑及与赖氨酸侧链结合的1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸钆结构,可在高表达ctsb的癌细胞内发生2-氰基苯并噻唑与d型半胱氨酸间点击缩合反应从而原位形成基于gd的纳米颗粒,提高了gd的胞内浓度,延长了gd的保留时间,实现了ctsb过表达肿瘤的t2加权磁共振对比度的增强,从而可用于ctsb相关癌症的早期诊断,具有良好的特异性;另外由于小分子在进入细胞后很容易被细胞排出,本发明的胞内构建的ctsb响应的t2型小分子磁共振造影剂,通过2-氰基苯并噻唑(cbt)与d型半胱氨酸(cys)间的点击缩合反应,利用小分子前体在细胞内组装成纳米结构的智能策略,结合了小分子和纳米结构互补的优势,小分子进入细胞后可转化为两亲性的寡聚体,进一步自组装成纳米结构,一方面具备小分子容易被细胞摄取的特点,另一方面也具备纳米结构很难被细胞泵出的特点。这种智能策略能有效延长小分子化合物在细胞内的滞留时间,提高其在细胞内的浓度,从而具有更好的成像或治疗效果。而且本发明采用的2-氰基苯并噻唑(cbt)与d型半胱氨酸(cys)间的点击缩合反应所触发的自组装过程具有条件温和、反应速度快且生物兼容性好的优点,同时可以在
ph、酶、以及还原型谷胱甘肽(gsh)等多种生物信号的控制下,根据特定需要进行特定的修饰,具有更广泛的应用范围。本发明填补了利用点击缩合反应原位形成基于钆的纳米结构以增强t2加权的磁共振肿瘤成像从而来诊断ctsb相关癌症方面的空白。
附图说明
[0021]
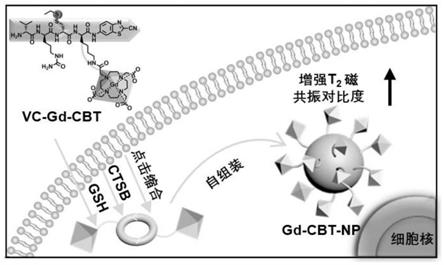
图1为本发明vc-gd-cbt的结构式以及vc-gd-cbt自组装形成的gd-cbt-nps增强了高磁场下(9.4t)肿瘤的t2磁共振对比度的示意图。
[0022]
图2为实施例1中合成的第一种纯化合物a的质谱(esi-ms)分析结果图。
[0023]
图3为实施例1中合成的第二种纯化合物b的质谱(esi-ms)分析结果图。
[0024]
图4为实施例1中合成的第三种纯化合物c的质谱(esi-ms)分析结果图。
[0025]
图5为实施例1中合成的第四种纯化合物d的质谱(esi-ms)分析结果图。
[0026]
图6为实施例1中合成的第五种纯化合物e的质谱(esi-ms)分析结果图。
[0027]
图7为实施例1中合成的第六种纯化合物f的质谱(esi-ms)分析结果图。
[0028]
图8为实施例1中合成的第七种纯化合物g的质谱(esi-ms)分析结果图。
[0029]
图9为实施例1中合成的第八种纯化合物h的质谱(esi-ms)分析结果图。
[0030]
图10为实施例1中合成的第八种纯化合物h的氢谱(1h nmr)分析结果图。
[0031]
图11为实施例1中合成的第八种纯化合物h的碳谱(
13
c nmr)分析结果图。
[0032]
图12为实施例1中合成的第九种纯化合物vc-gd-cbt的高分辨质谱(hr-maldi/ms)分析结果图。
[0033]
图13为实施例2中vc-gd-cbt在体外被ctsb酶切前后的高效液相色谱图。
[0034]
图14为实施例2中vc-gd-cbt被ctsb酶切然后发生cbt-cys点击缩合反应后形成的gd-cbt-dimer的高分辨质谱(hr-maldi/ms)分析结果图。
[0035]
图15为实施例2中形成的gd-cbt-dimer经过π-π自组装进一步形成的gd-cbt-nps的透射电镜图像。
[0036]
图16为实施例2中不同gd浓度下,gd-cbt-nps、vc-gd-cbt或gd-dtpa在ctsb的工作缓冲液中的横向弛豫率(1/t2)和纵向弛豫率(1/t1)。
[0037]
图17为实施例3中mda-mb-231细胞与vc-gd-cbt孵育8个小时以后的低倍率透射电镜图像和图a中白色矩形方框所圈区域的透射电子显微镜放大图。
[0038]
图18为实施例3中对照组inh.+vc-gd-cbt、对照组gd-dtpa和空白组mda-mb-231细胞的低倍透射电镜图像。
[0039]
图19为实施例3中不同gd浓度下,vc-gd-cbt组、inh.+vc-gd-cbt组和gd-dtpa组mda-mb-231细胞的横向弛豫率(1/t2)和纵向弛豫率(1/t1)。
[0040]
图20为实施例4中静脉注射vc-gd-cbt的mda-mb-231荷瘤小鼠(最上面一行)、先用ctsb抑制剂预处理然后再注射vc-gd-cbt的mda-mb-231荷瘤小鼠(中间一行)和注射gd-dtpa的mda-mb-231荷瘤小鼠(最下面一行)在0个小时(左列)和1.5个小时(右列)时候的t2加权冠状面mr图像。白色的圆圈表示肿瘤。
具体实施方式
[0041]
下面将结合附图通过实施例对本发明作进一步详细的说明。其中实施例1提供了
第一种纯化合物a,第二种纯化合物b,第三种纯化合物c,第四种纯化合物d,第五种纯化合物e,第六种纯化合物f,第七种纯化合物g,第八种纯化合物h,第九种纯化合物vc-gd-cbt的合成。实施例2中提供了ctsb引导的gd-cbt-nps的体外形成及增强了t2磁共振成像实验结果。实施例3中提供了细胞内形成的gd-cbt-nps增强了mda-mb-231细胞的t2磁共振成像实验结果。实施例4中提供了ctsb引导的gd-cbt-nps的形成增强了体内肿瘤的t2磁共振成像实验结果。
[0042]
实施例1:纯化合物a、b、c、d、e、f、g、h和vc-gd-cbt的合成
[0043]
本实施例具体介绍一种可在高表达ctsb的癌细胞内发生2-氰基苯并噻唑与d型半胱氨酸间点击缩合反应从而原位形成基于gd的纳米结构的小分子t2型磁共振造影剂的制备方法,共分九步完成,合成路线如下:
[0044][0045]
第一步:将2mmol(937mg)的氨基酸boc-lys(fmoc)-oh和4mmol(443μl)的4-甲基吗啉(mmp)溶于4ml的无水四氢呋喃(thf)中,冷却至0℃,加入2mmol(254μl)的氯甲酸异丁酯(ibcf),在0℃的条件下反应混合物搅拌一个小时。然后将1.67mmol的2-氰基-6-氨基苯并噻唑(cbt)溶液(溶剂为无水thf)加入到反应混合物中,先在0℃下进一步搅拌一个小时,然后在室温下继续搅拌过夜。经过高效液相色谱分离纯化,得到第一种纯化合物,命名为化合物a;
[0046]
第二步:将1.36mmol(848.5mg)的化合物a溶解在含有4ml二氯甲烷(dcm)、76ml三氟乙酸(tfa)和800μl三异丙基硅烷(tips)的混合溶液中(其中三氟乙酸和二氯甲烷的体积比为95:5,三异丙基硅烷的体积为溶液总体积的百分之一),室温下搅拌三个小时去除化合物a的叔丁氧羰基(boc)保护基团。经过高效液相色谱分离纯化,得到第二种纯化合物,命名为化合物b;
[0047]
第三步:将1.17mmol(433.7mg)的氨基酸boc-cys(set)-oh、1.17mmol(158.3mg)的1-羟基苯并三唑(hobt)、1.17mmol(444.3mg)的o-苯并三氮唑-四甲基脲六氟磷酸酯(hbtu)、1.17mmol(204μl)的n,n-二异丙基乙胺(dipea)和0.78mmol(410.2mg)的化合物b溶解在n,n-二甲基甲酰胺中,室温下搅拌12个小时。经过高效液相色谱分离纯化,得到第三种纯化合物,命名为化合物c;
[0048]
第四步:将0.54mmol(425.7mg)的化合物c溶解在含有2.5ml二氯甲烷(dcm)、
47.5ml三氟乙酸(tfa)和500μl三异丙基硅烷(tips)的混合溶液中,室温下搅拌三个小时去除化合物c的叔丁氧羰基(boc)保护基团。经过高效液相色谱分离纯化,得到第四种纯化合物,命名为化合物d;
[0049]
第五步:将0.62mmol(231.8mg)的氨基酸boc-val-cit-oh、0.87mmol(118mg)的1-羟基苯并三唑(hobt)、0.87mmol(330mg)的o-苯并三氮唑-四甲基脲六氟磷酸酯(hbtu)、0.87mmol(152μl)的n,n-二异丙基乙胺(dipea)和0.58mmol(395.9mg)的化合物d溶解在n,n-二甲基甲酰胺中,室温下搅拌24个小时。经过高效液相色谱分离纯化,得到第五种纯化合物,命名为化合物e;
[0050]
第六步:化合物e的n-芴甲氧羰酰基(fmoc)保护基团用含10%哌啶的n,n-二甲基甲酰胺溶液(0.6ml哌啶+5.4ml的n,n-二甲基甲酰胺)在0℃下搅拌反应10分钟,然后加入540μl三氟乙酸用于中和其中的碱。经过高效液相色谱分离纯化,得到第六种纯化合物,命名为化合物f;
[0051]
第七步:将0.38mmol(217.8mg)的三叔丁基1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸(dota(otbu)3)、0.38mmol(51.3mg)的1-羟基苯并三唑(hobt)和0.38mmol(144.1mg)的o-苯并三氮唑-四甲基脲六氟磷酸酯(hbtu)溶解于2毫升的n,n-二甲基甲酰胺中,随后加入0.38mmol(66μl)的n,n-二异丙基乙胺(dipea),室温下搅拌0.5个小时,然后将溶解于n,n-二甲基甲酰胺中的0.32mmol(260.6mg)的化合物f加入上述溶液中,室温下继续搅拌过夜。经过高效液相色谱分离纯化,得到第七种纯化合物,命名为化合物g;
[0052]
第八步:将0.19mmol(263.5mg)的化合物g溶解在含有1.5ml二氯甲烷(dcm)、28.5ml三氟乙酸(tfa)和300μl三异丙基硅烷(tips)的混合溶液中,室温下搅拌三个小时去除化合物g的叔丁氧羰基(boc)和氧叔丁基(otbu)保护基团。经过高效液相色谱分离纯化,得到第八种纯化合物,命名为化合物h;
[0053]
第九步:将0.043mmol(15.9mg)的氯化钆(gdcl3·
6h2o)溶解在水中,并用饱和碳酸钠溶液调节ph值为6.5-7.2,然后将其加入到0.02mmol(22.2mg)化合物h的n,n-二甲基甲酰胺溶液中,并将该混合溶液的ph值调节至6.5-7.2,在室温下搅拌反应3个小时。经过高效液相色谱分离纯化,即可得到目标化合物基于gd的小分子t2型造影剂vc-gd-cbt。
[0054]
图1为本发明vc-gd-cbt的结构式以及ctsb触发细胞内vc-gd-cbt自组装成gd-cbt-nps以增强高磁场下(9.4t)肿瘤的t2磁共振对比度的示意图。从图1可以看到当vc-gd-cbt进入过表达ctsb的癌细胞后,通过细胞内ctsb引导和cbt-cys点击缩合反应触发的自组装在癌细胞内原位形成gd纳米颗粒(gd-cbt-nps)。这一过程不仅提高了gd的胞内浓度和t2加权mr对比度,而且延长了gd的保留时间,导致t2加权图像上的信号强度明显低于通过ctsb抑制剂预处理的细胞的t2信号强度,说明vc-gd-cbt在ctsb过表达的癌细胞中原位自组装形成的gd纳米颗粒能够增强体内肿瘤的t2加权磁共振成像。
[0055]
采用赛默飞公司生产的finnigan lcq先进离子捕获质谱仪对纯化合物a、b、c、d、e、f、g、h进行电喷雾离子质谱数据采集分别得到如图2、图3、图4、图5、图6、图7、图8和图9的质谱图;采用德国布鲁克公司(bruker)布鲁克核磁软件解析纯化合物h得到如图10和图11所示的核磁共振谱图;采用布鲁克道尔顿飞行时间质谱仪对纯化合物vc-gd-cbt进行基质辅助激光解吸/电离质谱数据采集得到如图12的高分辨质谱图:
[0056]
图2是本实施例中合成的第一种纯化合物a的质谱图;图3是本实施例中合成的第
二乙二胺五乙酸造影剂)的t2和t1加权磁共振体模成像。得到了在不同gd浓度下,gd-cbt-nps、vc-gd-cbt或gd-dtpa在ctsb的工作缓冲液中的横向弛豫率(1/t2)和纵向弛豫率(1/t1),结果如图16所示,计算得到的gd-cbt-nps的r2值为16.01mm-1
s-1
,vc-gd-cbt的r2值7.01mm-1
s-1
,gd-dtpa的r2值为6.75mm-1
s-1
,表明体外ctsb引导的gd-cbt-nps的形成使得vc-gd-cbt的r2值提高了2.28倍。然而,gd-cbt-nps的r1值(4.28mm-1
s-1
)与vc-gd-cbt(4.68mm-1
s-1
)和gd-dtpa (5.48mm-1
s-1
)的r1值相似,表明vc-gd-cbt的纵向弛豫(r1)不受gd-cbt-nps形成的影响。此外,进一步计算得出gd-cbt-nps的r2/r1比值为3.74。这表明ctsb引导的gd-cbt-nps的形成大大缩短了周围水质子的横向弛豫时间(t2),导致t2磁共振信号增强,可在9.4t时作为t2主导的磁共振造影剂。
[0060]
实施例3:细胞内形成的gd-cbt-nps增强了mda-mb-231细胞的t2磁共振成像
[0061]
将mda-mb-231细胞分为四组:实验组vc-gd-cbt:mda-mb-231细胞与300μm的vc-gd-cbt共孵育8个小时;对照组inh.+vc-gd-cbt:2mm的ca-074-me(一种ctsb抑制剂)预处理1.5个小时以后,300μm vc-gd-cbt再与mda-mb-231细胞共孵育8个小时;对照组gd-dtpa:300μm gd-dtpa与mda-mb-231细胞共孵育8个小时;空白组为未经任何处理的细胞。然后,为了验证细胞内ctsb引导的gd-cbt-nps的形成,对上述四组细胞进行tem观察。图17为实施例3中mda-mb-231细胞与vc-gd-cbt孵育8个小时以后的低倍率透射电镜图像和图a中白色矩形方框所圈区域的透射电子显微镜放大图,由图17可以看到vc-gd-cbt实验组的细胞中存在大面积的gd-cbt-nps。同时,将图17a中白色矩形方框所圈区域进行放大,可以看到gd-cbt-nps自组装并在细胞内积累。图18为实施例3中对照组inh.+vc-gd-cbt、对照组gd-dtpa和空白组mda-mb-231细胞的低倍透射电镜图像,由图18可以看到在这三个对照组细胞中未发现gd-cbt-nps。然后稀释了mda-mb-231细胞样本并进行了t2加权磁共振体模成像。图19为实施例3中不同gd浓度下,vc-gd-cbt组、inh.+vc-gd-cbt组和gd-dtpa组mda-mb-231细胞的横向弛豫率(1/t2)和纵向弛豫率(1/t1),由图19可以看到,经计算得到的实验组vc-gd-cbt的r2值为21.12mm-1
s-1
,对照组inh.+vc-gd-cbt的r2值为7.33mm-1
s-1
,对照组gd-dtpa的r2值为6.96mm-1
s-1
,这表明细胞内ctsb引导的gd-cbt-nps的形成使vc-gd-cbt的r2值在9.4t时提高了2.88倍。三组细胞的r1值(vc-gd-cbt的r1值为5.86mm-1
s-1
,inh.+vc-gd-cbt的r1值为4.25mm-1
s-1
,gd-dtpa的r1值为5.56mm-1
s-1
)基本不变。另外,计算得到对照组inh.+vc-gd-cbt的r2/r1的比值为1.72,低于实验组vc-gd-cbt的r2/r1的比值3.60。以上这些实验结果证实了vc-gd-cbt可以通过ctsb引导的原位自组装有效地产生gd-cbt-nps,提高过表达ctsb的癌细胞的r2值和r2/r1的比值,从而可以用于在9.4t时成像细胞内ctsb的活性。
[0062]
实施例4:ctsb引导的gd-cbt-nps的形成增强了体内肿瘤的t2磁共振成像
[0063]
在每只balb/c裸鼠右大腿皮下移植500万过表达ctsb的mda-mb-231细胞。当肿瘤直径大小达到约7-10mm时,将裸鼠随机分为三组:实验组vc-gd-cbt中的小鼠静脉注射0.08mmol/kg的vc-gd-cbt;对照组inh.+vc-gd-cbt中的小鼠预先静脉注射0.25mmol/kg的ca-074-me(一种ctsb抑制剂),待作用1.5个小时后再静脉注射0.08mmol/kg的vc-gd-cbt;对照组gd-dtpa中的小鼠静脉注射0.08mmol/kg的gd-dtpa。在9.4t磁共振扫描仪上对三组小鼠进行动态t2和t1加权冠状面磁共振成像。图20为实施例4中静脉注射vc-gd-cbt的mda-mb-231荷瘤小鼠(最上面一行)、先用ctsb抑制剂预处理然后再注射vc-gd-cbt的mda-mb-231荷瘤小鼠(中间一行)和注射gd-dtpa的mda-mb-231荷瘤小鼠(最下面一行)在0个小时
(左列)和1.5个小时(右列)时候的t2加权冠状面mr图像。白色的圆圈表示肿瘤,由图20可以看到注射vc-gd-cbt的小鼠肿瘤的t2加权磁共振对比变化明显大于注射ctsb抑制剂ca-074-me和vc-gd-cbt的小鼠肿瘤以及注射gd-dtpa的小鼠肿瘤。表明vc-gd-cbt在ctsb引导下形成的gd-cbt-nps在9.4t时可以显著且特异性地提高ctsb过表达的肿瘤的t2磁共振对比度。
[0064]
以上数据表明,在9.4t时,ctsb引导的gd-cbt-nps的形成增加了gd在肿瘤部位的局部浓度和滞留时间,从而显著增强了体内过表达ctsb肿瘤的t2磁共振对比度。因此本发明设计的基于gd的小分子t2型造影剂vc-gd-cbt在未来临床应用的高磁场中有潜力作为一种有吸引力的造影剂用于t2加权磁共振成像诊断ctsb过表达的恶性肿瘤。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1