一种厄贝沙坦中间体的制备方法与流程
1.本发明属于药物化学领域,具体涉及一种厄贝沙坦中间体的制备方法。
背景技术:
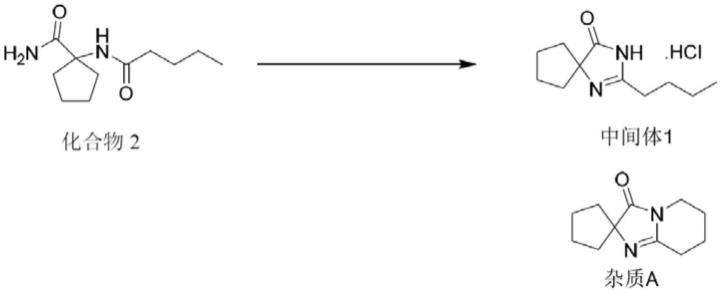
2.厄贝沙坦为血管紧张素ii(angiotensinv ii,ang ii)受体拮抗剂,能特异性地拮抗血管紧张素ii 1型受体(at1)。厄贝沙坦具有高效、长效、安全、可口服以及耐受性好等特点,并且有心、脑、肾保护作用,目前是市售主流降血压药物。厄贝沙坦的化学名称为2-丁基-3-{[2'-(1h-四唑-5-基)-[1,1'-联苯]-4-基]甲基}-1,3-二氮杂螺[4,4]壬-1-烯-4-酮,化学结构式如下:
[0003][0004]
现有技术中厄贝沙坦主要通过中间体1经过若干步骤获得,中间体1主要采用1-戊酰氨基环戊烷-1-甲酰胺化合物2在碱性条件下关环,而后酸化成盐获得,反应过程中产生一种杂质a,该杂质a在反应液中含量较高,且很难去除,不利于控制中间体1的质量,进而影响原料药厄贝沙坦的质量。常规技术手段采取3次以上的重结晶工序提纯至杂质a含量低于0.10%,多次结晶使得生产成本大幅上升。
[0005]
技术实现要素:
[0006]
针对现有技术的不足,本发明的目的是提供一种厄贝沙坦中间体1的制备方法,该方法可以有效降低反应液中杂质a的含量,获得杂质a含量极低的中间体1。本发明的厄贝沙坦中间体1的制备方法生产成本低,操作安全,易于工业应用。
[0007]
本发明的厄贝沙坦中间体1的制备方法包括以下步骤:
[0008]
将化合物2与碱、醇类溶剂混合,在加热温度下向混合液中通入氧气进行反应,反
应结束后,用酸调ph=8~9,然后减压浓缩,残留物用乙酸乙酯提取,乙酸乙酯层用氯化氢气体成盐,分离获得中间体1。
[0009][0010]
根据本发明的方法,其中所述醇类溶剂为甲醇、乙醇、异丙醇中的一种或多种,优选甲醇和/或异丙醇。
[0011]
根据本发明的方法,其中所述碱为氢氧化钠和/或氢氧化钾。
[0012]
根据本发明的方法,其中碱与化合物2的摩尔比为2~3:1,优选2.2:1。
[0013]
根据本发明的方法,其中通入氧气时加热温度为50~65℃,优选55~60℃。
[0014]
根据本发明的方法,其中通入氧气的流速控制在500~800ml/分钟。
[0015]
根据本发明的方法,其中通入氧气进行反应的反应时间为3~6小时。
[0016]
根据本发明的方法,其中反应结束后降至0~25℃,优选0~15℃,再用酸调ph=8~9。
[0017]
根据本发明的方法,其中调ph所使用的酸为磷酸、硫酸或盐酸,优选30%盐酸。
[0018]
根据本发明的方法,其中减压浓缩在45~65℃的温度下,优选50~60℃的温度下进行。
[0019]
根据本发明的方法,其中控制乙酸乙酯层的温度在0~25℃的温度范围内,通入氯化氢气体成盐,成盐时间为1.5~3小时,优选2小时。
[0020]
根据本发明的方法,其中,成盐后降温至0~10℃,优选0~5℃,保温0.5~1.5小时。
[0021]
有益效果
[0022]
本发明用通入氧气的方法合成厄贝沙坦中间体1,通过在反应过程中原位将可能的甲基自由基俘获并阻断,从而防止发生自由基环化反应生成杂质a,从而有效控制杂质a的含量低于0.10%,有利于后续厄贝沙坦原料药的质量控制,本发明的厄贝沙坦中间体1的制备方法操作简单,避免了中间体1的更多次结晶提纯操作,从而生产成本低,操作安全,易于工业应用。
附图说明
[0023]
图1是对比例制备的中间体1的hplc图谱。
[0024]
图2是实施例1制备的中间体1的hplc图谱。
[0025]
图3是实施例2制备的中间体1的hplc图谱。
[0026]
图4是实施例3制备的中间体1的hplc图谱。
[0027]
图5是实施例4制备的中间体1的hplc图谱。
[0028]
图6是实施例5制备的中间体1的hplc图谱。
具体实施方式
[0029]
下面结合具体实施例对本发明作进一步说明,但本发明的实施方式不限于此。
[0030]
本发明所用仪器为常规仪器,所使用的试剂为常规试剂。
[0031]
本发明所得中间体1的有关物质检测方法(hplc):
[0032]
色谱仪:agilent 1260
[0033]
溶液配制:
[0034]
流动相a:0.05%三氟乙酸水溶液
[0035]
流动相b:乙腈
[0036]
稀释液:乙腈/水(1:1,v/v)
[0037][0038]
对比例:中间体1的制备
[0039][0040]
在反应瓶中加入212g化合物2,88g氢氧化钠、1000g甲醇,搅拌溶解,升温至55~65℃保温反应3小时。反应结束后,降温至0~15℃,用30%盐酸调ph=8~9,然后控制55~60℃减压浓缩甲醇,残留物用500ml*3的乙酸乙酯提取四次,乙酸乙酯层控制0~25℃,通入氯化氢气体成盐2小时。而后降温至0~5℃,保温1小时,将所得晶体过滤,于70~80℃真空干燥12小时,得214g中间体1。收率93%,有关物质图谱见图1,其中杂质a含量0.8483%。
[0041]
实施例1:中间体1的制备
[0042]
[0043]
在反应瓶中加入212g化合物2,88g氢氧化钠、1000g甲醇,搅拌溶解,升温至55~60℃,将通气管插入混合液的液面以下,通过流量计调节氧气流速500ml/分钟,通入氧气保温反应3小时。反应结束后,降温至0~15℃,用30%盐酸调ph=8~9,然后控制50~60℃减压浓缩甲醇,残留物用500ml*3的乙酸乙酯提取两次,乙酸乙酯层控制0~25℃,通入氯化氢气体成盐2小时。而后降温至0~5℃,保温1小时,将所得晶体过滤,于70~80℃真空干燥12小时,得219g中间体1。收率95%,有关物质图谱见图2,其中杂质a含量0.0192%。
[0044]
实施例2:中间体1的制备
[0045][0046]
在反应瓶中加入212g化合物2,88g氢氧化钠、1000g甲醇,搅拌溶解,升温至55~60℃,将通气管插入混合液的液面以下,通过流量计调节氧气流速800ml/分钟,通入氧气保温反应3小时。反应结束后,降温至0~15℃,用30%盐酸调ph=8~9,然后控制50~60℃减压浓缩甲醇,残留物用500ml*3的乙酸乙酯提取两次,乙酸乙酯层控制0~25℃,通入氯化氢气体成盐2小时。而后降温至0~5℃,保温1小时,将所得晶体过滤,于70~80℃真空干燥12小时,得221g中间体1。收率96%,有关物质图谱见图3,其中杂质a含量0.0391%。
[0047]
实施例3:中间体1的制备
[0048][0049]
在反应瓶中加入212g化合物2,120g氢氧化钠、1000g甲醇,搅拌溶解,升温至55~60℃,将通气管插入混合液的液面以下,通过流量计调节氧气流速800ml/分钟,通入氧气保温反应3小时。反应结束后,降温至0~15℃,用30%盐酸调ph=8~9,然后控制50~60℃减压浓缩甲醇,残留物用500ml*3的乙酸乙酯提取两次,乙酸乙酯层控制0~25℃,通入氯化氢气体成盐2小时。而后降温至0~5℃,保温1小时,将所得晶体过滤,于70~80℃真空干燥12小时,得215g中间体1。收率93%,有关物质图谱见图4,其中杂质a含量0.0516%。
[0050]
实施例4:中间体1的制备
[0051][0052]
在反应瓶中加入212g化合物2,88g氢氧化钠、1000g甲醇,搅拌溶解,升温至55-65℃,将通气管插入混合液的液面以下,通过流量计调节氧气流速800ml/分钟,通入氧气保温
反应3小时。反应结束后,降温至0~15℃,用30%盐酸调ph=8~9,然后控制55-60℃减压浓缩甲醇,残留物用500ml*3的乙酸乙酯提取两次,乙酸乙酯层控制0~25℃,通入氯化氢气体成盐2小时。而后降温至0~5℃,保温1小时,将所得晶体过滤,于70~80℃真空干燥12小时,得212g中间体1。收率92%,有关物质图谱见图5,其中杂质a含量0.0725%。
[0053]
实施例5:中间体1的制备
[0054][0055]
在反应瓶中加入212g化合物2,88g氢氧化钠、1000g异丙醇,搅拌溶解,升温至55~60℃,将通气管插入混合液的液面以下,通过流量计调节氧气流速800ml/分钟,通入氧气保温反应3小时。反应结束后,降温至0~15℃,用30%盐酸调ph=8~9,然后控制50~60℃减压浓缩异丙醇,残留物用500ml*3的乙酸乙酯提取两次,乙酸乙酯层控制0~25℃,通入氯化氢气体成盐2小时。而后降温至0~5℃,保温1小时,将所得晶体过滤,于70~80℃真空干燥12小时,得217g中间体1。收率94%,有关物质图谱见图6,其中杂质a含量0.0350%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1