一种羧基功能化的时间分辨荧光微球及其制备方法与流程
1,4-丁二酮、4,4,4-三氟-1-苯基-1,3-丁二酮、1-(4-氯苯基)-4,4,4-三氟-1,3-丁二酮和4,4,4-三氟-1-(2-萘基)-1,3-丁二酮中的一种或多种;所述协同配体为4,7-二甲基-1,10-菲罗啉、2-腈基-1,10-邻菲啰啉、1,10-菲罗啉和3,4,7,8-四甲基-1,10-菲罗啉中的一种或多种;所述桥联配体为对苯二甲酸、邻苯二甲酸和间苯二甲酸中的一种或多种。
13.在本发明的一个实施例中,在步骤(1)中,采用带有强给电子基团的β-二酮类配体,在该基团影响下,二酮结构更容易向醇烯式结构转变,使其与稀土离子的配位能力更强。
14.在本发明的一个实施例中,在步骤(1)中,采用平面结构更大,刚性更强的协同配体,能够使整个荧光分子的刚性更强,从而减少分子内震动带来的能量损耗,提高荧光强度。
15.在本发明的一个实施例中,在步骤(1)中,铕离子与镧系金属离子的摩尔比为10:0.5-1:2。
16.在本发明的一个实施例中,在步骤(1)中,所述铕盐、β-二酮类配体、协同配体和桥联配体的摩尔比为1:6-10:1-3:1-3。
17.在本发明的一个实施例中,在步骤(1)中,所述酸性条件的ph为5-6;所述ph的调节剂为naoh的醇溶液或氨水。
18.在本发明的一个实施例中,所述naoh的醇溶液是质量分数为50%的氢氧化钠乙醇溶液。
19.在本发明的一个实施例中,在步骤(1)中,所述有机溶剂为甲苯、二氯甲烷、三氯甲烷、四氢呋喃中的一种或多种。
20.在本发明的一个实施例中,在步骤(2)中,所述丙烯酸类单体为2-苯基丙烯酸单体、丙烯酸和甲基丙烯酸中的一种或多种;所述引发剂为偶氮二异丁腈、过氧化二苯甲酰、过硫酸铵和过硫酸钾中的一种或多种;所述乳化剂为十二烷基硫酸钠、十二烷基苯磺酸钠和聚乙烯醇中的一种或多种。
21.在本发明的一个实施例中,在步骤(2)中,所述双核稀土配合物、苯乙烯单体、丙烯酸类单体、引发剂、聚乙烯吡咯烷酮、乳化剂的质量比为2-30:100:5-15:0.1-10:1-50:10-25。
22.在本发明的一个实施例中,在步骤(2)中,所述丙烯酸类单体在聚苯乙烯微球表面引入羧基,通过改变其加入量来改变聚苯乙烯微球表面的羧基含量。
23.在本发明的一个实施例中,在步骤(2)中,所述乳化剂可以改善苯乙烯微粒在反应体系中的分散性。
24.在本发明的一个实施例中,在步骤(2)中,所述反应的温度为65-75℃;反应的时间为10-14h。
25.在本发明的一个实施例中,在步骤(2)中,所述溶剂为水和/或乙醇。
26.本发明的第二个目的是提供一种所述的制备方法制备的羧基功能化的时间分辨荧光微球。
27.在本发明的一个实施例中,所述羧基功能化的时间分辨荧光微球的结构如下:
[0028][0029]
在本发明的一个实施例中,所述羧基功能化的时间分辨荧光微球的粒径为50nm-5μm。通过调节引发剂、苯乙烯来调节微球粒径、粒径分布。
[0030]
本发明的第三个目的是提供一种所述的羧基功能化的时间分辨荧光微球在免疫层析中的应用。
[0031]
本发明的技术方案相比现有技术具有以下优点:
[0032]
(1)本发明所述的制备方法将时间分辨荧光染料直接引入聚合体系中,使得时间分辨荧光染料直接进入微球内部,通过聚合将其包埋于聚合物内部。
[0033]
(2)本发明所述的制备方法在聚合阶段将染料直接包被于微球内部,无需考虑时间分辨荧光染料位阻效应,可以选用位阻更大,但荧光强度更强,无法应用于溶胀法的双核稀土配合物作为时间分辨荧光染料,从而使得时间分辨荧光微球的荧光强度更高。
[0034]
(3)本发明所述的制备方法无需单独制备羧基聚合物微球,无需使用有机溶剂对聚合物微球进行溶胀,从而简化了制备步骤,减少了对环境有害的有机溶剂使用。同时由于不需要使用有机溶剂对聚合微球溶胀,减少了微球变形风险,从而使得时间分辨荧光微球的粒径更均一,表面形貌更好,在医学临床检查、时间分辨免疫层析领域应用效果良好。
[0035]
(4)本发明所述的制备方法步骤简化,将聚苯乙烯微球制备与微球染色整合为一步完成,易于规模化生产。
附图说明
[0036]
为了使本发明的内容更容易被清楚地理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中:
[0037]
图1为本发明测试例1中的扫描电镜表征,其中,a为实施例1;b为对比例2。
[0038]
图2为本发明测试例2中的扫描电镜表征,其中,a为实施例2;b为对比例4。
[0039]
图3为本发明测试例3中的荧光光谱图。
[0040]
图4为本发明测试例4中的荧光光谱图。
具体实施方式
[0041]
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0042]
在本发明中,除非另有说明,双核稀土配合物的制备方法如下:
[0043]
(1)称取0.183g六水氯化铕、0.188g六水氯化镝、1.42g4,4,4-三氟-1-(4-三氟甲基苯基)-1,3-丁二酮、0.416g4,7-二甲基-1,10-菲罗啉,0.187g对苯二甲酸溶于50ml体积比1:1的乙醇、甲苯混合溶液;
[0044]
(2)逐滴滴加50%氢氧化钠溶液,调节ph约5,搅拌反应4h;
[0045]
(3)离心分离,洗涤,干燥,得到镝-铕双核稀土配合物。
[0046]
在本发明中,除非另有说明,单核稀土配合物的制备方法如下:
[0047]
(1)称取0.366g六水氯化铕、1.42g4,4,4-三氟-1-(4-三氟甲基苯基)-1,3-丁二酮、0.416g4,7-二甲基-1,10-菲罗啉,溶于50ml体积比1:1的乙醇、甲苯混合溶液;
[0048]
(2)逐滴滴加50%氢氧化钠溶液,调节ph约5,搅拌反应4h;
[0049]
(3)离心分离,洗涤,干燥,得到单核铕配合物。
[0050]
实施例1
[0051]
一种200nm羧基功能化的时间分辨荧光微球及其制备方法,具体包括以下步骤:
[0052]
(1)称取25g纯水、25g无水乙醇混合均匀,加入0.5gpvp、0.1gsds搅拌均匀;
[0053]
(2)向1g纯化后的苯乙烯单体中加入0.08g双核稀土配合物,充分搅拌溶解;
[0054]
(3)在500r/min搅拌下,将步骤(2)所得溶液加入步骤(1)所得溶液中,同时加入0.1g2-苯基丙烯酸单体,搅拌5min;
[0055]
(4)反应体系升温至40℃,在氮气吹扫下,反应2h,以除去二氯甲烷;
[0056]
(5)向反应体系中加入0.05g过硫酸铵升温至70℃,反应12h;
[0057]
(6)使用醇/水离心洗净产物数次,最后分散于纯水中,得到200nm羧基功能化的时间分辨荧光微球。
[0058]
实施例2
[0059]
一种300nm羧基功能化的时间分辨荧光微球及其制备方法,具体包括以下步骤:
[0060]
基本同实施例1,不同之处在于:
[0061]
步骤(5)所使用过硫酸铵量由原来的0.05g调整为0.03g。
[0062]
对比例1
[0063]
溶胀法制备200nm单核稀土配合物染色的羧基功能化时间分辨荧光微球,具体包括以下步骤:
[0064]
1)羧基功能化聚苯乙烯微球制备
[0065]
(1)称取25g纯水、25g无水乙醇混合均匀,加入0.5gpvp、0.1gsds搅拌均匀;
[0066]
(2)在500r/min搅拌下,向步骤(1)所得溶液中加入1g苯乙烯单体,同时加入0.1g2-苯基丙烯酸单体,搅拌5min;
[0067]
(3)向反应体系中加入0.05g过硫酸铵升温至70℃,反应12h;
[0068]
(4)使用醇/水离心洗净产物数次,最后分散于纯水中。
[0069]
2)溶胀法制备羧基功能化时间分辨荧光微球
[0070]
(1)取1g羧基功能化聚苯乙烯微球分散于30g纯水中,加入1gsds充分搅拌溶解;
[0071]
(2)取0.08g双核稀土配合物荧光染料溶于2ml二氯甲烷;
[0072]
(3)在150r/min搅拌转速下,将步骤(2)所得溶液滴加至步骤(1)所得溶液中;
[0073]
(4)超声振荡5min,使其充分乳化;
[0074]
(5)在150r/min搅拌转速、30℃温度条件下,使微球溶胀染色1h;
[0075]
(6)将反应体系升温至60℃,反应1h,挥发去除二氯甲烷;
[0076]
(7)反应结束后,冷却1h,使反应物降至室温,离心法洗净,使用乙醇和纯水分别洗净3次。
[0077]
对比例2
[0078]
基本同对比例1,不同之处在于溶胀法制备羧基功能化时间分辨荧光微球:
[0079]
步骤(2)所使用的双核核稀土配合物替换为单核稀土配合物。
[0080]
对比例3
[0081]
基本同对比例1,不同之处在于羧基功能化聚苯乙烯微球制备:
[0082]
步骤(3)所使用过硫酸铵量由原来的0.05g调整为0.03g。
[0083]
对比例4
[0084]
基本同对比例1,不同之处在于:
[0085]
羧基功能化聚苯乙烯微球制备:步骤(3)所使用过硫酸铵量由原来的0.05g调整为0.03g;
[0086]
溶胀法制备羧基功能化时间分辨荧光微球:步骤(2)所使用的双核核稀土配合物替换为单核稀土配合物。
[0087]
对比例5
[0088]
基本同实施例1,不同之处在于:
[0089]
步骤(2)所使用的双核稀土配合物替换为单核稀土配合物。
[0090]
对比例6
[0091]
基本同实施例1,不同之处在于:
[0092]
步骤(2)所使用的双核稀土配合物替换为单核稀土配合物;
[0093]
步骤(5)所使用过硫酸铵量由原来的0.05g调整为0.03g。
[0094]
测试例1
[0095]
对实施例1和对比例1的样品进行扫描电镜表征,结果如图1所示。从图1a可以看出,实施例1采用一步法制备的200nm时间分辨荧光微球,其样品表面光滑,粒径均一,单分散性好;从图1b可以看出,传统两步法制备的200nm时间分辨荧光微球,第二部染色过程中,由于溶胀剂的侵蚀,样品粒径均一性变差,粒径分布变宽。
[0096]
测试例2
[0097]
对实施例2和对比例3的样品进行扫描电镜表征,结果如图2所示。从图2a可以看出,采用一步法制备的300nm时间分辨荧光微球样品表面光滑,粒径均一,单分散性好;从图2b可以看出,由于制备过程中溶胀剂的侵蚀,传统两步法制备的300nm时间分辨荧光微球,样品粒径均一性变差,粒径分布变宽。
[0098]
测试例3
[0099]
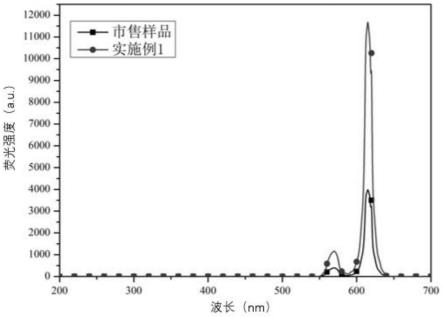
对市售样品(购自微测生物,210nmeu-时间分辨荧光微球)、实施例1、对比例1-2和5的样品进行荧光强度测试,荧光强度是使用荧光光度计,365nm激发条件下,测得的615nm
荧光强度值,结果如图3和表1所示:
[0100]
表1
[0101]
样品市售实施例1对比例1对比例2对比例5荧光强度40001180080051508100
[0102]
如图3所示,实施例1样品荧光强度远高于市售样品。
[0103]
如表1所示,与对比例5相比,实施例1选用荧光强度更高的双核稀土配合物作为荧光染料,所得样品荧光强度更高;对比例1由于采用溶胀法制备,位阻更大的双核稀土配合物染色效果较差,荧光强度较低。对比例5与对比例2相比,对比例5由于一步法制备的样品,染料被包埋于聚苯乙烯微球内部,无泄漏风险,而对比例2采用溶胀法制备,在相同染料用量条件下,染料损失大,且部分染料在洗净过程中泄露,因此荧光值相比于对比例5较低。
[0104]
测试例4
[0105]
对市售样品(购自微测生物,316nmeu-时间分辨荧光微球)、实施例2、对比例3-4和6的样品进行荧光强度测试,荧光强度是使用荧光光度计,365nm激发条件下,测得的615nm荧光强度值,结果如图4和表2所示:
[0106]
表2
[0107]
样品市售实施例2对比例6对比例3对比例4荧光强度60001650092408406820
[0108]
如图4所示,实施例2样品荧光强度远高于市售样品。
[0109]
如表1所示,与对比例6相比,实施例2选用荧光强度更高的双核稀土配合物作为荧光染料,所得样品荧光强度更高;对比例3由于采用溶胀法制备,位阻更大的双核稀土配合物染色效果较差,荧光强度较低。对比例6与对比例4相比,对比例6由于一步法制备的样品,染料被包埋于聚苯乙烯微球内部,无泄漏风险,而对比例4采用溶胀法制备,在相同染料用量条件下,染料损失大,且部分染料在洗净过程中泄露,因此荧光值相比于实施例2较低。
[0110]
测试例5
[0111]
对市售样品(购自微测生物,210nmeu-时间分辨荧光微球)、实施例1、对比例1-2和5的样品进行免疫层析测试,结果如表3所示:
[0112]
表3
[0113][0114][0115]
如表3所示,实施例1样品与市售样品相比,在免疫层析应用中有优异表现,其结果提高了近2倍。与实施例1相比,对比例5使用单核稀土配合物为染料,其裸球荧光值较低限制了其在免疫层析中的应用表现。与实施例1相比,对比例1样品,采用两步法制备,由于双
核配合物的位阻效应,其染色效果较差,产物荧光值低,应用结果较差。与实施例1相比,对比例2样品一方面使用的单核稀土配合物荧光值较双核配合物低,一方面微球粒径不够均一,表面腐蚀,导致其应用表现较实施例1差。
[0116]
测试例6
[0117]
对市售样品(购自微测生物,316nmeu-时间分辨荧光微球)、实施例2、对比例3-4和6的样品进行免疫层析测试,结果如表4所示:
[0118]
表4
[0119][0120]
如表4所示,其结果与200nm时间分辨荧光微球类似,实施例2样品采用双核稀土配合物为染料,一步法制备,其英勇表现良好,远高于市售样品。对比例6使用单核稀土配合物为染料,其裸球荧光值较低限制了其在免疫层析中的应用表现。与实施例2相比,对比例3样品,采用两步法制备,由于双核配合物的位阻效应,其染色效果较差,产物荧光值低,应用结果较差。与实施例2相比,对比例4样品一方面使用的单核稀土配合物荧光值较双核配合物低,一方面微球粒径不够均一,表面腐蚀,导致其应用表现较实施例2差。
[0121]
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1