美罗培南中间体MAP的制备方法与流程
美罗培南中间体map的制备方法
技术领域
1.本发明属于医药技术领域,具体涉及一种美罗培南中间体map的制备方法。
背景技术:
2.美罗培南类药物药用疗效好,对人副作用小,其应用前景广阔、附加值高。美罗培南中间体map主要用于美罗培南类药物的生产。
3.中国专利cn 101891743 a公开一种美罗培南中间体的合成方法,包括以下步骤:1) 将式(ii) 化合物在贵金属催化剂和助催化剂的作用下进行反应,得到含式(iv) 化合物的反应液a;2) 从反应液a得到含式(i) 化合物的溶液,反应路线如下:中国专利cn 113929684 a公开一种美罗培南中间体及其制备方法。美罗培南中间体的制备方法,包括:将氮杂双环类化合物、氯甲酸酯类化合物、第一有机碱和有机溶剂混合进行反应,得到美罗培南中间体;氮杂双环类化合物具有式ii所示结构,氯甲酸酯类化合物具有式iii所示结构,美罗培南中间体具有式i所示结构。所述美罗培南中间体的合成路线如下:由于上述专利中制备美罗培南中间体的生产工艺存在生产周期长、人工多、使用有机溶剂量大、废水量大、产品收率低等缺陷,所以导致产品利润率低,无法大规模生产。
4.目前,亟需提供一种工艺高效、环保、成本低、利润高、产品质量优势明显的美罗培南中间体map的制备方法。
技术实现要素:
5.本发明的目的是提供一种美罗培南中间体map的制备方法,从控制体系水分、提高绿色溶剂使用量、优化生产工序三个方面来提高收率、降低成本、减少三废产生,工艺高效、利润高,适合大规模工业化生产。
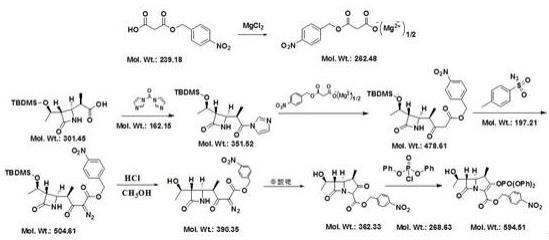
6.本发明所述的美罗培南中间体map的制备方法,包括如下步骤:(1)4bma酰化:以乙酸乙酯为溶剂,二异丙基乙胺为缚酸剂,二甲基吡啶为碱催化剂,4bma与羰基咪唑反应得到4bma酰化物溶液,4bma酰化物溶液脱水后得到4bma酰化液;(2)镁盐合成:以乙酸乙酯为溶剂,二异丙基乙胺为缚酸剂,单酸酯与无水氯化镁反应得到镁盐;(3)镁盐和4bma酰化物的反应:镁盐和4bma酰化液反应得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后得到脱水后的有机相;(4)重氮化:步骤(3)中得到的脱水后的有机相、对甲苯磺酰叠氮和三乙胺反应得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液;(5)脱保护基:向步骤(4)中得到的蒸馏液中加入甲醇,然后加入乙酸乙酯和盐酸进行反应得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液中加入正庚烷,离心,烘干,得到中间体;(6)环合:步骤(5)中得到的中间体与乙酸乙酯混合后,依次加入溴化锌和辛酸铑升温回流反应,得到环合产物;(7)缩合:环合产物中加入二氯甲烷、4-二甲氨基吡啶、氯代磷酸二苯酯和三乙胺反应得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液降温离心,烘干,得到美罗培南中间体map。
7.步骤(1)中4bma、羰基咪唑、二异丙基乙胺、二甲基吡啶与乙酸乙酯的质量比为1:0.5-0.6:0.5-0.7:0.05-0.07: 2-3。
8.步骤(1)中反应温度为15-22℃,反应时间为25-35min。
9.步骤(1)中脱水是采用无水硫酸镁或分子筛进行脱水,4bma与分子筛的质量比为1: 0.5-1.0,4bma与无水硫酸镁的质量比为1:0.1-0.6。
10.步骤(1)中4bma酰化液中的水分<0.1%。
11.步骤(2)中单酸酯、无水氯化镁、二异丙基乙胺与乙酸乙酯的质量比为1:0.21-0.28:0.5-0.6:2.4-2.8。
12.步骤(2)中反应温度为30-35℃,反应时间为30-40min。
13.步骤(3)中4bma酰化液与镁盐的质量比为1:0.8-1.5。
14.步骤(3)中反应温度为30-35℃,反应时间为15-20h。
15.步骤(3)中洗涤是反应液依次进行酸洗、碱洗和盐洗,其中,酸洗是反应液中加入浓度为3-6%的盐酸溶液进行酸洗,搅拌,静置,分层,分去酸洗水相,有机相保留;碱洗是酸洗结束后,有机相中加入5%碳酸钾溶液进行碱洗,搅拌,静置,分层,分去碱洗水相,有机相保留;盐洗是碱洗结束后,有机相中加入10%氯化钠溶液进行盐洗,搅拌,静置,分层,分去盐洗水相,有机相保留。
16.4bma、盐酸溶液、碳酸钾溶液与氯化钠溶液的质量比为1:8-12:8-12:2-5。
17.步骤(3)中反萃是反应液洗涤产生的水相采用乙酸乙酯进行反萃,4bma与乙酸乙酯的质量比为1:2.0-4.0。
18.反萃的具体步骤如下:
乙酸乙酯中加入酸洗水相,搅拌,静置,分层,水相分走,有机相中加入碱洗水相,搅拌,静置,分层,水相分走,有机相中加入盐洗水相,搅拌,静置,分层,水相分走,有机相与洗涤后得到的有机相合并。
19.步骤(3)中脱水是采用无水硫酸镁进行脱水,脱水时间为30-60min。
20.步骤(1)中的4bma、步骤(4)中的对甲苯磺酰叠氮与步骤(4)中的三乙胺的质量比为1:1:0.28-0.3。
21.步骤(4)中反应温度为0-5℃,反应时间为1-2h。
22.步骤(4)中洗涤是反应液依次进行酸洗、一次盐洗和二次盐洗,其中,酸洗是反应液中加入浓度为1.7-2.5%的盐酸溶液进行酸洗,搅拌,静置,分层,分去酸洗水相,有机相保留;一次盐洗是酸洗结束后,有机相中加入10%氯化钠溶液进行一次盐洗,搅拌,静置,分层,分去一次盐洗水相,有机相保留;二次盐洗是一次盐洗结束后,有机相中加入10%氯化钠溶液进行二次盐洗,搅拌,静置,分层,分去二次盐洗水相,有机相保留。
23.4bma、盐酸溶液、一次盐洗中的氯化钠溶液与二次盐洗中的氯化钠溶液的质量比为1:8-12:8-12:8-12。
24.反萃是反应液洗涤产生的水相采用乙酸乙酯进行反萃,4bma与乙酸乙酯的质量比为1:2.0-4.0。
25.反萃的具体步骤如下:乙酸乙酯中加入酸洗水相,搅拌,静置,分层,水相分走,有机相中加入一次盐洗水相,搅拌,静置,分层,水相分走,有机相中加入二次盐洗水相,搅拌,静置,分层,水相分走,有机相与洗涤后得到的有机相合并。
26.步骤(4)中脱水是采用无水硫酸镁进行脱水,脱水时间为30-60min。
27.步骤(4)中蒸馏为负压蒸馏。
28.步骤(4)中对甲苯磺酰叠氮优选为对甲苯磺酰叠氮的乙酸乙酯溶液(含50wt.%乙酸乙酯)。由于对甲苯磺酰叠氮属于叠氮化物,而叠氮化物在高温、撞击等条件下易爆炸,所以将对甲苯磺酰叠氮溶于乙酸乙酯中稳定性更好,加入三乙胺(活化α氢)中,对甲苯磺酰叠氮在碱性条件下更加稳定不易分解。
29.步骤(5)中甲醇的温度为0-5℃。
30.步骤(1)中的4bma、步骤(5)中的甲醇与步骤(5)中的盐酸的质量比为1:0.9-1.1:1.9-2.0。
31.步骤(5)中反应温度为15-18℃,反应时间为2-3.5小时。
32.步骤(5)中盐酸为20%稀盐酸。
33.步骤(5)中乙酸乙酯与盐酸的质量比为100:60-70。
34.步骤(5)中洗涤是反应液依次进行一次盐洗和二次盐洗,其中,一次盐洗是反应液中加入一次盐洗溶液进行一次盐洗,搅拌,静置,分层,分去一次盐洗水相,有机相保留,一次盐洗溶液为10%碳酸钾溶液和20%磷酸氢二钠溶液的混合溶液;二次盐洗是一次盐洗结束后,有机相中加入二次盐洗溶液进行二次盐洗,搅拌,静
置,分层,分去二次盐洗水相,有机相保留,二次盐洗溶液为10%氯化钠溶液和20%磷酸氢二钠溶液的混合溶液。
35.4bma、一次盐洗溶液与二次盐洗溶液的质量比为1:6-10:6-10。
36.反萃是反应液洗涤产生的水相采用乙酸乙酯进行反萃。
37.反萃的具体步骤如下:乙酸乙酯中加入一次盐洗水相,搅拌,静置,分层,水相分走,有机相中加入二次盐洗水相,搅拌,静置,分层,水相分走,有机相与洗涤后得到的有机相合并。
38.步骤(5)中脱水是采用无水硫酸镁进行脱水,脱水时间为30-60min。
39.步骤(5)中蒸馏为负压蒸馏,蒸馏温度为55-65℃步骤(1)中的4bma与步骤(5)中的正庚烷的质量比为1:3.2-3.6。
40.步骤(6)中乙酸乙酯、中间体、溴化锌与辛酸铑的质量比为160-210:100:0.8-1.1:0.8-1.1。
41.步骤(6)中溴化锌的加入温度为30-35℃,辛酸铑的加入温度为60-65℃,升温回流反应温度为72-76℃,升温回流反应时间为40-70min。
42.步骤(1)中的4bma、步骤(7)中的氯代磷酸二苯酯、步骤(7)中的三乙胺与步骤(7)中的4-二甲氨基吡啶的质量比为1:0.3-0.4:0.4-0.5:0.05-0.07,步骤(5)中的中间体与步骤(7)中的二氯甲烷的重量比为1:1.5-3。
43.步骤(7)中反应温度为-18—-12℃,反应时间为2-3h。
44.步骤(7)中洗涤是反应液依次进行一次盐洗和二次盐洗,其中,一次盐洗是反应液中加入一次盐洗溶液进行一次盐洗,搅拌,静置,分层,分去一次盐洗水相,有机相保留,一次盐洗溶液为10%磷酸二氢钾溶液和10%氯化钠溶液的混合溶液;二次盐洗是一次盐洗结束后,有机相中加入二次盐洗溶液进行二次盐洗,搅拌,静置,分层,分去二次盐洗水相,有机相保留,二次盐洗溶液为10%磷酸二氢钾溶液和10%磷酸氢二钠溶液的混合溶液。
45.4bma、一次盐洗溶液与二次盐洗溶液的质量比为1:5-8:5-8。
46.反萃是反应液洗涤产生的水相采用二氯甲烷进行反萃。
47.反萃的具体步骤如下:二氯甲烷中加入一次盐洗水相,搅拌,静置,分层,水相分走,有机相中加入二次盐洗水相,搅拌,静置,分层,水相分走,有机相与洗涤后得到的有机相合并。
48.步骤(7)中脱水是采用无水硫酸镁进行脱水,脱水时间为30-60min。
49.步骤(7)中蒸馏为负压蒸馏,蒸馏温度为55-65℃。
50.步骤(7)中降温至0-5℃。
51.本发明的反应方程式如下:
本发明所述的美罗培南中间体map的制备方法,包括如下具体步骤:(1)4bma酰化:反应瓶内加入乙酸乙酯、4bma、 羰基咪唑(cdi)和二甲基吡啶,内温15-22℃溶解,滴加二异丙基乙胺(dipea),保温反应25-35min,反应完毕后得到4bma酰化物溶液,4bma酰化物溶液经无水硫酸镁或分子筛脱水后得到4bma酰化液(水分<0.1%,水分越低越好);(2)镁盐合成:反应瓶内加入乙酸乙酯、单酸酯和无水氯化镁,内温30-35℃溶解,滴加二异丙基乙胺(dipea),保温30-40min,得到镁盐;(3)镁盐和4bma酰化物的反应:镁盐和4bma酰化液反应15-20h得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后得到脱水后的有机相,其中,洗涤:反应液依次进行酸洗、碱洗和盐洗;酸洗:加入浓度为3-6%盐酸进行酸洗,搅拌,静置,分层,分去酸洗水相,有机相保留;碱洗:酸洗结束后,有机相中加入5%碳酸钾溶液进行碱洗,搅拌,静置,分层,分去碱洗水相,有机相保留;盐洗:碱洗结束后,有机相中加入10%氯化钠溶液进行盐洗,搅拌,静置,分层,分去盐洗水相,有机相保留。
52.反萃:反应瓶内提前加入乙酸乙酯,先转入酸洗结束后的酸洗水相,搅拌,静置,分层,水相分走,有机相在反应瓶内,再转入碱洗结束后的碱洗水相,搅拌,静置,分层,水相分走,有机相在反应瓶内,再转入盐洗结束后的盐洗水相,搅拌,静置,分层,水相分走,有机相在反应瓶内;将三种洗涤水相分别用乙酸乙酯反萃,将水中有机相回收至乙酸乙酯相中,再将乙酸乙酯相同洗涤工序盐洗结束后的有机相合并,进入脱水工序。
53.脱水:有机相中加入无水硫酸镁搅拌30-60min,过滤,得到脱水后的有机相。
54.(4)重氮化:步骤(3)中得到的脱水后的有机相、对甲苯磺酰叠氮和三乙胺内温0-5℃反应1-2h得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液;其中,洗涤:反应液依次进行酸洗、一次盐洗和二次盐洗;酸洗:加入浓度为1.7-2.5%盐酸进行酸洗,搅拌,静置,分层,分去酸洗水相,有机相保留;酸洗结束后,有机相中加入10%氯化钠溶液进行一次盐洗,搅拌,静置,分层,分去一次盐洗水相,有机相保留;一次盐洗结束后,有机相中加入10%氯化钠溶液进行二次盐洗,搅拌,静置,分层,分去二次盐洗水相,有机相保留。
55.反萃:反应瓶内提前加入乙酸乙酯,先转入酸洗结束后的酸洗水相,搅拌,静置,分层,水相分走,有机相在反应瓶内,再转入一次盐洗结束后的一次盐洗水相,搅拌,静置,分
层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌,静置,分层,水相分走,有机相在反应瓶内;将三种洗涤水相分别用乙酸乙酯反萃,将水中有机相回收至乙酸乙酯相中,再将乙酸乙酯相同洗涤工序盐洗结束后的有机相合并,进入脱水工序。
56.脱水:有机相加入无水硫酸镁搅拌30-60min,过滤。
57.蒸馏:负压蒸馏(将溶剂乙酸乙酯蒸出套用至洗涤及反萃工序),补加乙酸乙酯,蒸四次(水分<0.25%)(将溶剂乙酸乙酯蒸出套用至洗涤及反萃工序),得到蒸馏液。
58.(5)脱保护基:向步骤(4)中得到的蒸馏液中加入预冷的甲醇(作用:1、体系析出晶体,2、洗涤有机相溶解的杂质对甲苯磺酰胺),然后加入乙酸乙酯和浓度为20%盐酸15-18℃反应2-3.5小时(酸性强酯键断裂分解)得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液中加入正庚烷,离心,烘干,得到中间体;其中,洗涤:配置一次盐洗溶液:10%碳酸钾溶液和20%磷酸氢二钠溶液的混合溶液,将反应液转入配置好的一次盐洗溶液中进行一次盐洗,搅拌,静置,分层,一次盐洗水相进入反萃工序,有机相进行二次盐洗;配置二次盐洗溶液:10%氯化钠溶液和20%磷酸氢二钠溶液的混合溶液,有机相转入配置好的二次盐洗溶液中进行二次盐洗,搅拌,静置,分层,二次盐洗水相进入反萃工序,有机相保留。
59.反萃:反应瓶内提前加入乙酸乙酯,先转入一次盐洗结束后的一次盐洗水相,搅拌,静置,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌,静置,分层,水相分走,有机相同洗涤工序盐洗结束后的有机相合并,进入脱水工序。
60.脱水:有机相中加入无水硫酸镁搅拌30-60min,过滤。
61.蒸馏:55-65℃负压蒸馏(将溶剂乙酸乙酯蒸出套用至洗涤及反萃工序),补加乙酸乙酯,蒸四次(水分<0.25%,将溶剂乙酸乙酯蒸出套用至洗涤及反萃工序)。
62.滴加正庚烷析晶,离心,烘干得中间体,离心母液为正庚烷母液,暂存于-2—2℃储罐内析出沉降后,沉降套用至析晶岗位,可以在不套用母液基础上提高收率1-3%,上层正庚烷母液经精馏后得到纯度99.0-99.8%的正庚烷套用至本岗位。
63.(6)环合:步骤(5)中得到的中间体与乙酸乙酯混合后,30-35℃加入溴化锌,60-65℃加入辛酸铑,升温回流反应40-70min,蒸出ea,得到环合产物。
64.(7)缩合:环合产物中加入二氯甲烷、4-二甲氨基吡啶(dmap)、氯代磷酸二苯酯和三乙胺-18—-12℃反应2-3h得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液降温离心,烘干,得到美罗培南中间体map;其中,洗涤:配置一次盐洗溶液:10%磷酸二氢钾溶液和10%氯化钠溶液的混合溶液,将反应液转入配置好的一次盐洗溶液中进行一次盐洗,搅拌,静置,分层,一次盐洗水相进入反萃工序,有机相进行二次盐洗。配置二次盐洗溶液:10%磷酸二氢钾溶液和10%磷酸氢二钠溶液的混合溶液,将有机相转入配置好的二次盐洗溶液中进行二次盐洗,搅拌,静置,分层,二次盐洗水相进入反萃工序,有机相保留。
65.反萃:反应瓶内提前加入二氯甲烷,先转入一次盐洗结束后的一次盐洗水相,搅拌,静置,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌,静置,分层,水相分走,有机相同洗涤工序盐洗结束后的有机相合并,进入脱水工序。
66.脱水:有机相中加入无水硫酸镁搅拌30-60min,过滤。
67.蒸馏:55-65℃负压蒸馏,先蒸馏出二氯甲烷套用至萃取工序,再蒸馏出乙酸乙酯
套用至脱保护基工序,补加乙酸乙酯,蒸四次(水分<0.25%),乙酸乙酯套用至脱保护基工序。
68.降温至0-5℃离心、烘干得美罗培南中间体map产品;离心母液为乙酸乙酯母液,暂存于-2—2℃储罐内析出沉降后,沉降套用至析晶岗位,可以提高收率1-3%,上层乙酸乙酯母液经精馏后得到纯度99.0-99.8%乙酸乙酯回收套用。
69.本发明的主要优势及特点如下:一、反应体系水分控制:美罗培南中间体map分子量较大,含有多个酯基、多元环等易发生分解导致产品收率低,所以需要对反应体系水分进行控制。
70.(1)反应体系硫酸镁脱水:重氮化、脱保护基等步骤中反应液加入无水硫酸镁循环脱水,保证体系水分在0.1%以下,防止中间体酯基的水解,及原料对甲苯磺酰叠氮、辛酸铑、氯代磷酸二苯酯的水解,防止副反应发生,提高收率至95%以上,纯度达到99.6%以上。
71.(2)多次高温蒸馏控制反应体系水分:脱保护基工序及缩合工序将溶剂乙酸乙酯内温55-65℃蒸出后,分别补加4次新乙酸乙酯55-65℃蒸馏,将体系内乙酸乙酯蒸掉,并控制水分<0.25%析晶。
72.二、绿色溶剂使用:4bma酰化、镁盐合成、重氮化、脱保护基、环合工序母液体系为单一绿色溶剂乙酸乙酯。脱保护基工序析晶使用甲醇和正庚烷析晶,从源头上采用了绿色溶剂,实现了有机溶剂的全部回收套用,回收率可达到90-95%,降低了生产成本。
73.三、优化生产工序:(1)4bma((3s,4s)-3-((r)-1-(叔丁基二甲基硅氧基)乙基)-4((r)-1-甲酰乙基)-2-氮杂环丁酮)酰化:使用二异丙基乙胺为缚酸剂生产盐酸盐,是反应向顺方向进行。使用二甲基吡啶为碱催化剂的优点是活化羧基。
74.(2)镁盐和4bma酰化物的反应:采用3-6%的盐酸酸洗,再碳酸钾碱洗、氯化钠盐洗,保证中间体不被分解。使用弱酸、弱碱、盐溶液进行洗涤,洗涤液ph较弱不会造成长链的断键。
75.(3)重氮化:采用含50%乙酸乙酯的对甲苯磺酰叠氮,溶于三乙胺(活化α氢)中,可以保证工序更加安全,转化率更高。
76.(4)重氮化后蒸出乙酸乙酯使用甲醇析晶,溶解后不离心甩料,一锅法滴加20%盐酸,低温反应进行脱保护基。
77.(5)脱保护基多次补加乙酸乙酯蒸馏后,使用正庚烷析晶,离心得到中间体。正庚烷母液沉降套用至正庚烷析晶岗位可以在不套用母液基础上提高收率1-3%,蒸馏正庚烷纯度99.0-99.8%后循环套用。
78.(6)环合工序:加入溴化锌,防止五元环甲基的异构化;60-65℃加入辛酸铑是采用高温环合反应;采用高温回流采出的方式提高催化剂单位反应液浓度,提高收率。
79.(7)缩合工序:使用dmap(4-二甲氨基吡啶)作为碱性催化剂,dmap较吡啶碱性强、选择性更高,在低温15-20℃就可以加成反应。乙酸乙酯母液沉降套用至析晶岗位可以提高收率1-3%,蒸馏乙酸乙酯纯度99.0-99.8%后循环套用。
80.本发明的有益效果如下:(1)成本方面:通过工序水分的控制、反萃、母液套用及各工序的优化,产品收率提
高至95%以上,生产成本大大降低,利润相当可观,工艺操作简单,单元设备少,可进行连续化工业生产。
81.(2)质量方面:洗涤过程都是采用稀酸、稀碱、稀盐进行处理,有效的防止中间体的分解。对蒸馏水分的控制,可以保证析晶过程水分低,产品在高温蒸馏过程中不被分解。所得产品外观及质量指标都有较大提高,产品反式、单一杂质都会大大降低,产品质量明显提升,纯度可达到99.6%以上。
82.(3)环保方面:使用乙酸乙酯、甲醇、正庚烷等绿色溶剂进行生产,实现了有机溶剂的全部回收套用,回收率可达到90-95%,降低了生产成本。
具体实施方式
83.以下结合实施例对本发明做进一步描述。
84.实施例1(1)4bma酰化:反应瓶内加入80g乙酸乙酯、36g 4bma、20gcdi(羰基咪唑)和2g二甲基吡啶,内温20℃溶解,滴加dipea(二异丙基乙胺)20g,保温30min,反应完毕后得到4bma酰化物溶液,4bma酰化物溶液中加入无水硫酸镁10g脱水30min,过滤,得到4bma酰化液。
85.(2)镁盐合成:反应瓶内加入100g乙酸乙酯、40g单酸酯和10g无水氯化镁,内温30℃溶解,滴加dipea(二异丙基乙胺)20g,保温40min,得到镁盐。
86.(3)镁盐和4bma酰化物的反应:步骤(1)中得到的4bma酰化液和步骤(2)中得到的镁盐30℃反应20h得到反应液,中控测4bma酰化残留,合格后反应液经洗涤和反萃后得到有机相,有机相脱水后得到脱水后的有机相,其中,洗涤:反应液依次进行酸洗、碱洗和盐洗;酸洗:加入5%盐酸300g,搅拌30min,静置30min,分层,酸洗水相转入另一反应瓶反萃(提前加入100g乙酸乙酯),有机相保留瓶内进行碱洗;碱洗:加入5%碳酸钾溶液350g,搅拌30min,静置30min,分层,碱洗水相反萃,有机相保留瓶内进行盐洗;盐洗:加入10%氯化钠100g,搅拌30min,静置30min,分层,盐洗水相反萃,有机相保留。
87.反萃:反应瓶内加入100g乙酸乙酯,再加入酸洗水相,搅拌30min,静置30min,分层,水相分走,有机相暂存瓶内,再转入碱洗水相搅拌30min,静置30min,分层,水相分走,有机相暂存瓶内,再转入盐洗水相搅拌30min,静置30min,分层,水相分走,有机相同洗涤有机相合并。
88.脱水:加入无水硫酸镁10g搅拌30min过滤。
89.(4)重氮化:步骤(3)中得到的脱水后的有机相、对甲苯磺酰叠氮36g和三乙胺10.8g内温0℃反应2h得到反应液。中控残留合格后,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液;其中,洗涤:反应液依次进行酸洗、一次盐洗和二次盐洗;酸洗:加入2%盐酸300g,搅拌30min,静置30min,分层得有机相,酸洗水相反萃;一次盐洗:酸洗结束后,有机相中加入10%氯化钠溶液300g,搅拌30min,静置30min,分层得有机相,一次盐洗水相反萃;二次盐洗:一次盐洗结束后,有机相中加入10%氯化钠溶液350g,搅拌30min,静置30min,分层得有机相,二次盐洗水相反萃。
90.反萃:反应瓶内提前加入100g乙酸乙酯,先转入酸洗结束后的酸洗水相,搅拌
30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入一次盐洗结束后的一次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内;有机相同洗涤有机相合并。
91.脱水:加入无水硫酸镁10g搅拌30min过滤。
92.蒸馏:
①
负压蒸至内温60℃;
②
补加乙酸乙酯50g,蒸馏;
③
重复步骤
②
三次,得到蒸馏液。
93.(5)脱保护基:向步骤(4)中得到的蒸馏液中加入预冷的0℃甲醇36g,然后加入乙酸乙酯100g和浓度为20%盐酸70g 16℃反应2小时得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液中加入正庚烷,离心,烘干,得到中间体;其中,洗涤:配置一次盐洗溶液:10%碳酸钾溶液和20%磷酸氢二钠溶液的混合溶液250g,将反应液转入配置好的一次盐洗溶液中进行一次盐洗,搅拌30min,静置30min,分层,一次盐洗水相进入反萃工序,有机相进行二次盐洗;配置二次盐洗溶液:10%氯化钠溶液和20%磷酸氢二钠溶液的混合溶液300g,有机相转入配置好的二次盐洗溶液中进行二次盐洗,搅拌30min,静置30min,分层,二次盐洗水相进入反萃工序,有机相保留。
94.反萃:反应瓶内提前加入100g乙酸乙酯,先转入一次盐洗结束后的一次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相同洗涤工序盐洗结束后的有机相合并,进入脱水工序。
95.脱水:有机相中加入无水硫酸镁10g搅拌30min,过滤。
96.蒸馏:
①
负压蒸至内温65℃;
②
补加乙酸乙酯50g,蒸馏;
③
重复步骤
②
三次,内温不超65℃,得到蒸馏液。
97.滴加正庚烷120g析晶,离心,烘干得中间体55g,纯度99.2%,离心母液为正庚烷母液,暂存于-2—2℃储罐内析出沉降后,沉降套用至析晶岗位,上层正庚烷母液经精馏后得到纯度99.0%的正庚烷循环套用。
98.(6)环合:步骤(5)中得到的中间体55g与乙酸乙酯100g混合后,35℃加入溴化锌0.5g,65℃加入辛酸铑0.5g,升温回流反应60min,升温回流反应温度为75℃,蒸出ea50g,得到环合产物。
99.(7)缩合:环合产物中加入二氯甲烷100g、4-二甲氨基吡啶2.0g、氯代磷酸二苯酯11g和三乙胺15g
ꢀ‑
15℃反应3h得到反应液,中控测残留,合格后反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液降温离心,烘干,得到美罗培南中间体map;其中,洗涤:配置一次盐洗溶液:10%磷酸二氢钾溶液和10%氯化钠溶液的混合溶液200g,将反应液转入配置好的一次盐洗溶液中进行一次盐洗,搅拌30min,静置30min,分层,一次盐洗水相进入反萃工序,有机相进行二次盐洗。配置二次盐洗溶液:10%磷酸二氢钾溶液和10%磷酸氢二钠溶液的混合溶液200g,将有机相转入配置好的二次盐洗溶液中进行二次盐洗,搅拌30min,静置30min,分层,二次盐洗水相进入反萃工序,有机相保留。
100.反萃:反应瓶内提前加入二氯甲烷50g,先转入一次盐洗结束后的一次盐洗水相,
搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相同洗涤工序盐洗结束后的有机相合并,进入脱水工序。
101.脱水:有机相中加入无水硫酸镁10g搅拌30min,过滤。
102.蒸馏:
①
负压蒸至内温60℃;
②
补加乙酸乙酯50g,蒸馏;
③
重复步骤
②
三次,得到蒸馏液。
103.降温至0℃离心、烘干得美罗培南中间体map产品35g,以4bma计算质量收率97%,纯度99.6%;离心母液为乙酸乙酯母液,暂存于-2—2℃储罐内析出沉降后,沉降套用至析晶岗位,上层乙酸乙酯母液经精馏后得到纯度99.3%乙酸乙酯回收套用。
104.实施例2(1)4bma酰化:反应瓶内加入150g乙酸乙酯、70g 4bma、42gcdi(羰基咪唑)和4g二甲基吡啶,内温15℃溶解,滴加dipea(二异丙基乙胺)36g,保温25min,反应完毕后得到4bma酰化物溶液,4bma酰化物溶液中加入无水硫酸镁10g脱水30min,过滤,得到4bma酰化液。
105.(2)镁盐合成:反应瓶内加入200g乙酸乙酯、82g单酸酯和18g无水氯化镁,内温32℃溶解,滴加dipea(二异丙基乙胺)42g,保温35min,得到镁盐。
106.(3)镁盐和4bma酰化物的反应:步骤(1)中得到的4bma酰化液和步骤(2)中得到的镁盐32℃反应18h得到反应液,中控测4bma酰化残留,合格后反应液经洗涤和反萃后得到有机相,有机相脱水后得到脱水后的有机相,其中,洗涤:反应液依次进行酸洗、碱洗和盐洗;酸洗:加入5%盐酸600g,搅拌30min,静置30min,分层,酸洗水相转入另一反应瓶反萃(提前加入200g乙酸乙酯),有机相保留瓶内进行碱洗;碱洗:加入5%碳酸钾溶液600g,搅拌30min,静置30min,分层,碱洗水相反萃,有机相保留瓶内进行盐洗;盐洗:加入10%氯化钠200g,搅拌30min,静置30min,分层,盐洗水相反萃,有机相保留。
107.反萃:反应瓶内加入200g乙酸乙酯,再加入酸洗水相,搅拌30min,静置30min,分层,水相分走,有机相暂存瓶内,再转入碱洗水相搅拌30min,静置30min,分层,水相分走,有机相暂存瓶内,再转入盐洗水相搅拌30min,静置30min,分层,水相分走,有机相同洗涤有机相合并。
108.脱水:加入无水硫酸镁10g搅拌30min过滤。
109.(4)重氮化:步骤(3)中得到的脱水后的有机相、对甲苯磺酰叠氮70g和三乙胺20g内温2℃反应1.5h得到反应液。中控残留合格后,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液;其中,洗涤:反应液依次进行酸洗、一次盐洗和二次盐洗;酸洗:加入2%盐酸600g,搅拌30min,静置30min,分层得有机相,酸洗水相反萃;一次盐洗:酸洗结束后,有机相中加入10%氯化钠溶液700g,搅拌30min,静置30min,分层得有机相,一次盐洗水相反萃;二次盐洗:一次盐洗结束后,有机相中加入10%氯化钠溶液700g,搅拌30min,静置30min,分层得有机相,二次盐洗水相反萃。
110.反萃:反应瓶内提前加入200g乙酸乙酯,先转入酸洗结束后的酸洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入一次盐洗结束后的一次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后
的二次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内;有机相同洗涤有机相合并。
111.脱水:加入无水硫酸镁20g搅拌30min过滤。
112.蒸馏:
①
负压蒸至内温60℃;
②
补加乙酸乙酯100g,蒸馏;
③
重复步骤
②
三次,得到蒸馏液。
113.(5)脱保护基:向步骤(4)中得到的蒸馏液中加入预冷的2℃甲醇72g,然后加入乙酸乙酯200g和浓度为20%盐酸140g 18℃反应2.5小时得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液中加入正庚烷,离心,烘干,得到中间体;其中,洗涤:配置一次盐洗溶液:10%碳酸钾溶液和20%磷酸氢二钠溶液的混合溶液600g,将反应液转入配置好的一次盐洗溶液中进行一次盐洗,搅拌30min,静置30min,分层,一次盐洗水相进入反萃工序,有机相进行二次盐洗;配置二次盐洗溶液:10%氯化钠溶液和20%磷酸氢二钠溶液的混合溶液650g,有机相转入配置好的二次盐洗溶液中进行二次盐洗,搅拌30min,静置30min,分层,二次盐洗水相进入反萃工序,有机相保留。
114.反萃:反应瓶内提前加入100g乙酸乙酯,先转入一次盐洗结束后的一次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相同洗涤工序盐洗结束后的有机相合并,进入脱水工序。
115.脱水:有机相中加入无水硫酸镁20g搅拌30min,过滤。
116.蒸馏:
①
负压蒸至内温60℃;
②
补加乙酸乙酯100g,蒸馏;
③
重复步骤
②
三次,得到蒸馏液。
117.滴加正庚烷250g析晶,离心,烘干得中间体120g,纯度99.3%,离心母液为正庚烷母液,暂存于-2—2℃储罐内析出沉降后,沉降套用至析晶岗位,上层正庚烷母液经精馏后得到纯度99.2%的正庚烷循环套用。
118.(6)环合:步骤(5)中得到的中间体120g与乙酸乙酯200g混合后,32℃加入溴化锌1.0g,62℃加入辛酸铑1.0g,升温回流反应40min,升温回流反应温度为76℃,蒸出ea100g,得到环合产物。
119.(7)缩合:环合产物中加入二氯甲烷200g、4-二甲氨基吡啶4.2g、氯代磷酸二苯酯23g和三乙胺31g
ꢀ‑
12℃反应2h得到反应液,中控测残留,合格后反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液降温离心,烘干,得到美罗培南中间体map;其中,洗涤:配置一次盐洗溶液:10%磷酸二氢钾溶液和10%氯化钠溶液的混合溶液450g,将反应液转入配置好的一次盐洗溶液中进行一次盐洗,搅拌30min,静置30min,分层,一次盐洗水相进入反萃工序,有机相进行二次盐洗。配置二次盐洗溶液:10%磷酸二氢钾溶液和10%磷酸氢二钠溶液的混合溶液450g,将有机相转入配置好的二次盐洗溶液中进行二次盐洗,搅拌30min,静置30min,分层,二次盐洗水相进入反萃工序,有机相保留。
120.反萃:反应瓶内提前加入二氯甲烷80g,先转入一次盐洗结束后的一次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相同洗涤工序盐洗结束后的有机相
合并,进入脱水工序。
121.脱水:有机相中加入无水硫酸镁20g搅拌30min,过滤。
122.蒸馏:
①
负压蒸至内温60℃;
②
补加乙酸乙酯100g,蒸馏;
③
重复步骤
②
三次,得到蒸馏液。
123.降温至2℃离心、烘干得美罗培南中间体map产品68g,以4bma计算质量收率97%,纯度99.7%;离心母液为乙酸乙酯母液,暂存于-2—2℃储罐内析出沉降后,沉降套用至析晶岗位,上层乙酸乙酯母液经精馏后得到纯度99.1%乙酸乙酯回收套用。
124.实施例3(1)4bma酰化:反应瓶内加入100g乙酸乙酯、40g 4bma、23gcdi(羰基咪唑)和2.8g二甲基吡啶,内温22℃溶解,滴加dipea(二异丙基乙胺)28g,保温35min,反应完毕后得到4bma酰化物溶液,4bma酰化物溶液中加入无水硫酸镁10g脱水30min,过滤,得到4bma酰化液。
125.(2)镁盐合成:反应瓶内加入120g乙酸乙酯、44g单酸酯和12g无水氯化镁,内温35℃溶解,滴加dipea(二异丙基乙胺)25g,保温30min,得到镁盐。
126.(3)镁盐和4bma酰化物的反应:步骤(1)中得到的4bma酰化液和步骤(2)中得到的镁盐35℃反应15h得到反应液,中控测4bma酰化残留,合格后反应液经洗涤和反萃后得到有机相,有机相脱水后得到脱水后的有机相,其中,洗涤:反应液依次进行酸洗、碱洗和盐洗;酸洗:加入5%盐酸400g,搅拌30min,静置30min,分层,酸洗水相转入另一反应瓶反萃(提前加入100g乙酸乙酯),有机相保留瓶内进行碱洗;碱洗:加入5%碳酸钾溶液450g,搅拌30min,静置30min,分层,碱洗水相反萃,有机相保留瓶内进行盐洗;盐洗:加入10%氯化钠100g,搅拌30min,静置30min,分层,盐洗水相反萃,有机相保留。
127.反萃:反应瓶内加入100g乙酸乙酯,再加入酸洗水相,搅拌30min,静置30min,分层,水相分走,有机相暂存瓶内,再转入碱洗水相搅拌30min,静置30min,分层,水相分走,有机相暂存瓶内,再转入盐洗水相搅拌30min,静置30min,分层,水相分走,有机相同洗涤有机相合并。
128.脱水:加入无水硫酸镁10g搅拌30min过滤。
129.(4)重氮化:步骤(3)中得到的脱水后的有机相、对甲苯磺酰叠氮40g和三乙胺12g内温5℃反应1h得到反应液。中控残留合格后,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液;其中,洗涤:反应液依次进行酸洗、一次盐洗和二次盐洗;酸洗:加入2%盐酸400g,搅拌30min,静置30min,分层得有机相,酸洗水相反萃;一次盐洗:酸洗结束后,有机相中加入10%氯化钠溶液400g,搅拌30min,静置30min,分层得有机相,一次盐洗水相反萃;二次盐洗:一次盐洗结束后,有机相中加入10%氯化钠溶液350g,搅拌30min,静置30min,分层得有机相,二次盐洗水相反萃。
130.反萃:反应瓶内提前加入100g乙酸乙酯,先转入酸洗结束后的酸洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入一次盐洗结束后的一次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内;有机相同洗
涤有机相合并。
131.脱水:加入无水硫酸镁10g搅拌30min过滤。
132.蒸馏:
①
负压蒸至内温60℃;
②
补加乙酸乙酯52g,蒸馏;
③
重复步骤
②
三次,得到蒸馏液。
133.(5)脱保护基:向步骤(4)中得到的蒸馏液中加入预冷的5℃甲醇38g,然后加入乙酸乙酯130g和浓度为20%盐酸78g 15℃反应3.5小时得到反应液,反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液中加入正庚烷,离心,烘干,得到中间体;其中,洗涤:配置一次盐洗溶液:10%碳酸钾溶液和20%磷酸氢二钠溶液的混合溶液250g,将反应液转入配置好的一次盐洗溶液中进行一次盐洗,搅拌30min,静置30min,分层,一次盐洗水相进入反萃工序,有机相进行二次盐洗;配置二次盐洗溶液:10%氯化钠溶液和20%磷酸氢二钠溶液的混合溶液350g,有机相转入配置好的二次盐洗溶液中进行二次盐洗,搅拌30min,静置30min,分层,二次盐洗水相进入反萃工序,有机相保留。
134.反萃:反应瓶内提前加入100g乙酸乙酯,先转入一次盐洗结束后的一次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相同洗涤工序盐洗结束后的有机相合并,进入脱水工序。
135.脱水:有机相中加入无水硫酸镁10g搅拌30min,过滤。
136.蒸馏:
①
负压蒸至内温60℃;
②
补加乙酸乙酯53g,蒸馏;
③
重复步骤
②
三次,得到蒸馏液。
137.滴加正庚烷130g析晶,离心,烘干得中间体58g,纯度99.8%,离心母液为正庚烷母液,暂存于-2—2℃储罐内析出沉降后,沉降套用至析晶岗位,上层正庚烷母液经精馏后得到纯度99.1%的正庚烷循环套用。
138.(6)环合:步骤(5)中得到的中间体58g与乙酸乙酯120g混合后,30℃加入溴化锌0.6g,60℃加入辛酸铑0.6g,升温回流反应70min,升温回流反应温度为72℃,蒸出ea60g,得到环合产物。
139.(7)缩合:环合产物中加入二氯甲烷120g、4-二甲氨基吡啶2.8g、氯代磷酸二苯酯12g和三乙胺18g
ꢀ‑
18℃反应2.5h得到反应液,中控测残留,合格后反应液经洗涤和反萃后得到有机相,有机相脱水后蒸馏,得到蒸馏液,蒸馏液降温离心,烘干,得到美罗培南中间体map;其中,洗涤:配置一次盐洗溶液:10%磷酸二氢钾溶液和10%氯化钠溶液的混合溶液250g,将反应液转入配置好的一次盐洗溶液中进行一次盐洗,搅拌30min,静置30min,分层,一次盐洗水相进入反萃工序,有机相进行二次盐洗。配置二次盐洗溶液:10%磷酸二氢钾溶液和10%磷酸氢二钠溶液的混合溶液250g,将有机相转入配置好的二次盐洗溶液中进行二次盐洗,搅拌30min,静置30min,分层,二次盐洗水相进入反萃工序,有机相保留。
140.反萃:反应瓶内提前加入二氯甲烷60g,先转入一次盐洗结束后的一次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相在反应瓶内,再转入二次盐洗结束后的二次盐洗水相,搅拌30min,静置30min,分层,水相分走,有机相同洗涤工序盐洗结束后的有机相合并,进入脱水工序。
141.脱水:有机相中加入无水硫酸镁20g搅拌30min,过滤。
142.蒸馏:
①
负压蒸至内温60℃;
②
补加乙酸乙酯55g,蒸馏;
③
重复步骤
②
三次,得到蒸馏液。
143.降温至0℃离心、烘干得美罗培南中间体map产品38g,以4bma计算质量收率95%,纯度99.8%;离心母液为乙酸乙酯母液,暂存于-2—2℃储罐内析出沉降后,沉降套用至析晶岗位,上层乙酸乙酯母液经精馏后得到纯度99.2%乙酸乙酯回收套用。
144.对比例1(1)4bma酰化:反应瓶内加入200g二氯甲烷、50g 4bma和28gcdi(羰基咪唑),内温25℃溶解,保温反应8h,得到4bma酰化液。
145.(2)镁盐合成:反应瓶内加入220g二氯甲烷、50g单酸酯和15g无水氯化镁,内温30℃溶解,滴加三乙胺25g,保温反应30min,得到镁盐。
146.(3)镁盐和4bma酰化物的反应:步骤(1)中得到的4bma酰化液和步骤(2)中得到的镁盐30℃反应30h得到反应液,中控测4bma酰化残留,合格后反应液进行酸洗;酸洗:加入31%盐酸80g,搅拌30min,静置30min,分层,得到有机相。
147.(4)重氮化:步骤(3)中得到的有机相和对甲苯磺酰叠氮50g内温50℃反应2h,负压蒸至内温60℃蒸出溶剂,30℃滴加预冷的0℃异丙醇50g析晶得到湿品48g。
148.(5)脱保护基:200g丙酮、100g水和步骤(4)得到的湿品48g混合后,滴加31%盐酸78g,负压蒸至内温60℃,离心,烘干得中间体37g,纯度94.8%。
149.(6)环合:步骤(5)中得到的中间体37g与乙酸乙酯120g混合后,65℃加入辛酸铑0.6g,升温回流反应1h,得到环合产物。
150.(7)缩合:内温25℃环合产物中加入二氯甲烷120g、氯代磷酸二苯酯12g和三乙胺18g保温反应3h,中控测残留,合格后进行后续蒸馏:负压蒸至内温60℃,降至15℃离心烘干得map43g,以4bma计算质量收率86%,纯度95.8%。
151.对比例2各步均无脱水步骤,其它同实施例1,得到美罗培南中间体map产品32g,以4bma计算质量收率89%,纯度96.5%。
152.对比例3步骤(1)中不加入二异丙基乙胺和二甲基吡啶,其它同实施例1,得到美罗培南中间体map产品33.5g,以4bma计算质量收率93%,纯度98.9%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1