一种2-氟-4-溴-6-甲基苯甲酸甲酯的制备方法与流程
1.本发明涉及医药化工技术领域,尤其是涉及一种2-氟-4-溴-6-甲基苯甲酸甲酯的制备方法。
背景技术:
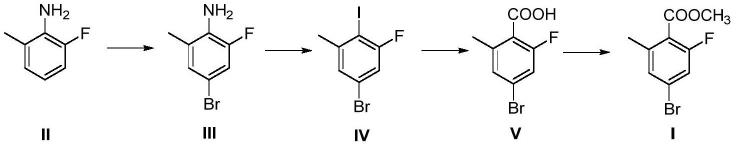
2.2-氟-4-溴-6-甲基苯甲酸甲酯是一类关键的医药中间体,wo2021/7477公开了如下合成2-氟-4-溴-6-甲基苯甲酸甲酯化合物i的路线:
[0003][0004]
上述合成方法中,在合成2-氟-4-溴-6-甲基苯甲酸甲酯化合物i的过程中用2-氟-4-溴-6-甲基苯甲酸为起始原料,该物料价格非常贵,不利于成本的控制。
技术实现要素:
[0005]
针对现有技术存在的上述问题,本发明申请人提供了一种2-氟-4-溴-6-甲基苯甲酸甲酯的制备方法。本发明制备方法的成本低,利于产业化生产。
[0006]
本发明的技术方案如下:
[0007]
本发明的第一个目的是提供一种2-氟-4-溴-6-甲基苯甲酸甲酯的制备方法,所述制备方法按照下述合成路线进行:
[0008][0009]
具体步骤为:
[0010]
(1)以2-氟-6-甲基苯胺为起始原料,与n-溴代丁二酰亚胺反应生成化合物iii;
[0011]
(2)化合物iii与亚硝酸钠生成重氮盐,在碘化钾、铜粉条件下发生桑德迈尔反应生成化合物iv;
[0012]
(3)化合物iv与镁生成格氏试剂,在二氧化碳条件下反应生成化合物v;
[0013]
(4)化合物v在浓硫酸催化下甲酯化生成化合物i;即所述2-氟-4-溴-6-甲基苯甲酸甲酯。
[0014]
在本发明的一个实施例中,步骤(1)中,反应溶剂为n,n-二甲基甲酰胺;所述2-氟-6-甲基苯胺与n-溴代丁二酰亚胺的摩尔比为1:1.1~1.3;反应条件为:温度为10~30℃,反应时间为1~3h。
[0015]
在本发明的一个实施例中,步骤(2)中,所述化合物iii与亚硝酸钠的摩尔比为1:1.1~1.3;重氮化反应条件为:温度为10~30℃,反应时间为1~3h。
[0016]
在本发明的一个实施例中,重氮化反应中溶剂为浓硫酸,浓硫酸的质量浓度为98%。
[0017]
在本发明的一个实施例中,步骤(2)中,所述碘化钾与化合物iii的摩尔比为1.1~1.3:1;铜粉与化合物iii的摩尔比为0.05:1;所述桑德麦尔反应的条件为:温度为10~30℃,反应时间为1~3h。
[0018]
在本发明的一个实施例中,步骤(3)中,反应溶剂为四氢呋喃、乙醚、甲基四氢呋喃中的一种或多种;所述化合物iv与镁、二氧化碳的摩尔比1:1.0~1.2:1.2~2.0。
[0019]
在本发明的一个实施例中,步骤(3)中,化合物iv与镁反应的条件为:温度为40~65℃,反应时间为2~6h。
[0020]
在本发明的一个实施例中,步骤(3)中,格氏试剂与二氧化碳反应的条件为:温度为0-10℃,反应时间为1~2h。
[0021]
在本发明的一个实施例中,步骤(4)中,溶剂为甲醇;所述化合物v与浓硫酸的摩尔比为1:0.01~0.05;反应条件为:温度为50~65℃,反应时间为3~6h。
[0022]
在本发明的一个实施例中,步骤(4)中,浓硫酸的质量浓度为98%;
[0023]
本发明有益的技术效果在于:
[0024]
本发明以廉价的2-氟-6-甲基苯胺为起始原料的全合成路线过程中成本低廉,易处理,且各步骤中最低的收率也达85%,总收率也达71%左右;且相比较用2-氟-4-溴-6-甲基苯胺(化合物ii)直接重氮化上氰基再酯化而言,避免了氰化钠、氰化亚铜等剧毒品的使用,对环境更加友好,更利于工业化生产。
附图说明
[0025]
图1为实施例1所得2-氟-4-溴-6-甲基苯甲酸甲酯的1h-nmr图谱;
[0026]
图2为实施例1所得2-氟-4-溴-6-甲基苯甲酸甲酯的气相色谱图。
具体实施方式
[0027]
下面结合附图和实施例,对本发明进行具体描述。
[0028]
实施例1
[0029]
一种2-氟-4-溴-6-甲基苯甲酸甲酯的制备方法,所述制备方法包括如下步骤:
[0030]
(1)化合物iii的合成
[0031]
向反应瓶中加入125g(1mol)2-氟-6-甲基苯胺,300ml n,n-二甲基甲酰胺,分批加入194.7g(1.1mol)n-溴代丁二酰亚胺,加完升温至30℃搅拌3小时,tlc跟踪反应直至2-氟-6-甲基苯胺反应完全;加水淬灭过滤烘干,得192.9g的化合物iii,摩尔收率:95%。
[0032]
(2)化合物iv的合成
[0033]
向反应瓶中加入300ml 98%硫酸,降温至30℃,分批加入53.1g(0.77mol)亚硝酸钠,再缓慢加入142.1g(0.7mol)化合物iii,控制反应温度30℃搅拌1小时,tlc点板跟踪反应直至化合物iii反应完全,制得重氮液;
[0034]
取另外一个反应瓶,依次加入500ml水、127.8g(0.77mol)碘化钾、2.24g(35mmol)铜粉,控温30℃抽入上述重氮液,控制反应温度30℃搅拌3小时,tlc点板跟踪反应直至上述重氮化合物反应完全,加二氯乙烷分层,水层二氯乙烷萃取,有机层浓缩,甲醇结晶得
182.4g化合物iv,摩尔收率:83%。
[0035]
(3)化合物v的合成
[0036]
向反应瓶中加入1000ml四氢呋喃,14.4g(600mmol)镁,升温至65℃,将157g(500mmol)化合物iv溶解在300ml四氢呋喃中缓慢滴加,滴完回流2小时至镁屑基本消失,gc跟踪反应直至化合物iv反应完全;降温至0℃,通26.4g(600mmol)二氧化碳,通完0℃保温搅拌2小时,tlc点板跟踪反应至反应完全,加1000ml饱和氯化铵水溶液淬灭反应,加入1000ml乙酸乙酯分层,饱和氯化钠洗涤,有机层干燥浓缩,甲苯结晶得98.6g化合物v,摩尔收率:85%。
[0037]
(4)化合物i的合成
[0038]
向反应瓶中加入23.2g(0.1mol)化合物v,100ml甲醇,0.1g(1mmol)浓硫酸,加完升温至65℃搅拌6小时,tlc跟踪反应直至化合物v反应完全;浓缩,加水打浆,固体过滤烘干得24.6g的化合物i,摩尔收率:100%。
[0039]
图1为2-氟-4-溴-6-甲基苯甲酸甲酯化合物i的氢谱,1h-nmr(400mhz,dmso-d6)δ:7.55~7.53(m,1h,arh),7.46(s,1h,arh),3.88(s,3h,och3),2.33(s,3h,ch3)。
[0040]
图2为2-氟-4-溴-6-甲基苯甲酸甲酯化合物i气相色谱图,可以看出2-氟-4-溴-6-甲基苯甲酸甲酯化合物i的纯度大于98%。
[0041]
实施例2
[0042]
一种2-氟-4-溴-6-甲基苯甲酸甲酯的制备方法,所述制备方法包括如下步骤:
[0043]
(1)化合物iii的合成
[0044]
向反应瓶中加入125g(1mol)2-氟-6-甲基苯胺,300mln,n-二甲基甲酰胺,分批加入212.4g(1.2mol)n-溴代丁二酰亚胺,加完升温至20℃搅拌2小时,tlc跟踪反应直至2-氟-6-甲基苯胺反应完全;加水淬灭过滤烘干,得194.9g的化合物iii,摩尔收率:96%。
[0045]
(2)化合物iv的合成
[0046]
向反应瓶中加入300ml 98%硫酸,降温至20℃,分批加入58g(0.84mol)亚硝酸钠,再缓慢加入142.1g(0.7mol)化合物iii,控制反应温度20℃搅拌2小时,tlc点板跟踪反应直至化合物iii反应完全,制得重氮液;
[0047]
取另外一个反应瓶,依次加入500ml水、139.4g(0.84mol)碘化钾、2.24g(35mmol)铜粉,控温20℃抽入上述重氮液,控制反应温度20℃搅拌2小时,tlc点板跟踪反应直至上述重氮化合物反应完全,加二氯乙烷分层,水层二氯乙烷萃取,有机层浓缩,甲醇结晶得186.8g化合物iv,摩尔收率:85%。
[0048]
(3)化合物v的合成
[0049]
向反应瓶中加入1000ml四氢呋喃,13.2g(550mmol)镁,升温至50℃,将157g(500mmol)化合物iv溶解在300ml四氢呋喃中缓慢滴加,滴完50℃搅拌4小时至镁屑基本消失,gc跟踪反应直至化合物iv反应完全;降温至-5℃,通33g(750mmol)二氧化碳,通完-5℃保温搅拌1.5小时,tlc点板跟踪反应至反应完全,加1000ml饱和氯化铵水溶液淬灭反应,加入1000ml乙酸乙酯分层,饱和氯化钠洗涤,有机层干燥浓缩,甲苯结晶得100.9g化合物v,摩尔收率:87%。
[0050]
(4)化合物i的合成
[0051]
向反应瓶中加入23.2g(0.1mol)化合物v,100ml甲醇,0.3g(3mmol)浓硫酸,加完升
温至55℃搅拌4小时,tlc跟踪反应直至化合物v反应完全;浓缩,加水打浆,固体过滤烘干得24.1g的化合物i,摩尔收率:98%。
[0052]
实施例3
[0053]
一种2-氟-4-溴-6-甲基苯甲酸甲酯的制备方法,所述制备方法包括如下步骤:
[0054]
(1)化合物iii的合成
[0055]
向反应瓶中加入125g(1mol)2-氟-6-甲基苯胺,300mln,n-二甲基甲酰胺,分批加入230.1g(1.3mol)n-溴代丁二酰亚胺,加完降温至10℃搅拌1小时,tlc跟踪反应直至2-氟-6-甲基苯胺反应完全;加水淬灭过滤烘干,得194.9g的化合物iii,摩尔收率:96%。
[0056]
(2)化合物iv的合成
[0057]
向反应瓶中加入300ml 98%硫酸,降温至10℃,分批加入62.8g(0.91mol)亚硝酸钠,再缓慢加入142.1g(0.7mol)化合物iii,控制反应温度10℃搅拌3小时,tlc点板跟踪反应直至化合物iii反应完全,制得重氮液;
[0058]
取另外一个反应瓶,依次加入500ml水、151g(0.91mol)碘化钾、2.24g(35mmol)铜粉,控温10℃抽入上述重氮液,控制反应温度10℃搅拌1小时,tlc点板跟踪反应直至上述重氮化合物反应完全,加二氯乙烷分层,水层二氯乙烷萃取,有机层浓缩,甲醇结晶得186.8g化合物iv,摩尔收率:85%。
[0059]
(3)化合物v的合成
[0060]
向反应瓶中加入1000ml四氢呋喃,12g(500mmol)镁,升温至40℃,将157g(500mmol)化合物iv溶解在300ml四氢呋喃中缓慢滴加,滴完40℃搅拌6小时至镁屑基本消失,gc跟踪反应直至化合物iv反应完全;降温至-10℃,通44g(1mol)二氧化碳,通完-10℃保温搅拌1小时,tlc点板跟踪反应至反应完全,加1000ml饱和氯化铵水溶液淬灭反应,加入1000ml乙酸乙酯分层,饱和氯化钠洗涤,有机层干燥浓缩,甲苯结晶得100.9g化合物v,摩尔收率:87%。
[0061]
(4)化合物i的合成
[0062]
向反应瓶中加入23.2g(0.1mol)化合物v,100ml甲醇,0.5g(5mmol)浓硫酸,加完升温至50℃搅拌3小时,tlc跟踪反应直至化合物v反应完全;浓缩,加水打浆,固体过滤烘干得24.1g的化合物i,摩尔收率:98%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1