一种F-18标记的羟基呋喃手性纯衍生物及其制备方法和应用
一种f-18标记的羟基呋喃手性纯衍生物及其制备方法和应用
技术领域
1.本发明属于药物合成技术领域,具体涉及新型的放射性核素标记探针及其制备方法,尤其涉及一种f-18标记的羟基呋喃手性纯衍生物及其制备方法和应用。
背景技术:
2.l-抗坏血酸(aa)被认为具有抗肿瘤作用并进行临床研究,但是并非对所有肿瘤都有效,需要寻求精准筛选工具甄别出对aa敏感的肿瘤。分子成像技术可以提供一种在细胞或者亚细胞水平有效监测生物分子的非入侵性方法,是一种可视化的精准筛选工具。在现有的分子成像技术中,正电子断层发射成像(pet)被认为是一种理想的工具。近年来人们应用放射性核素(
18
f、
125
i、
131
i、
11
c、
14
c)标记技术研究了与aa结构类似的相关化合物,
18
f标记的aa结构类似物则是很好的pet分子探针。
3.rumsey等人的研究表明,aa及其类似物在细胞中装运的结构要求之一是五元还原环的c-4位具有s-绝对构型,而c-2和c-3位没有被取代。通过对5-o-(4-[
125
i]碘苄基)-l-抗环血酸研究发现,在aa上的c-5位引入一个大的取代基,如碘苄氧基,其化合物并不能很好保持aa本身在体内特有的分布特性。而在aa的c-6位引入
18
f、
131
i等放射性同位素,显像显示其在啮齿动物的肾上腺等组织器官中显示出如预期的分布,因此表明c-6位点引入的取代基化合物适合作为放射性示踪剂。由于6-卤代-l抗坏血酸在原子坐标、键长和角度、氢坐标、各向异性和各向同性位移参数等方面与天然aa高度相似。放射化学合成的
18
f标记的6-卤代-l-抗坏血酸为可视化研究aa在体内、体外的作用过程提供生化功能均与aa高度类似的正电子示踪剂。
[0004]
与aa类似的正电子示踪剂 [
18
f]6-f-抗坏血酸([
18
f]dfa)的合成方法国外已有报道,其标记方法存在标记时间长(90 min),效率不高(4.9-15.6%)的缺陷。另外,文献显示已报到的分子探针[
18
f]dfa分子结构中c-6位的手性并未分开,表明[
18
f]dfa是具有多种构型的混合物,因而其生物性能与手性纯的抗坏血酸也有不同。文献表明[
18
f]dfa标记产率较低,且在小鼠体内生物性能一般,故而并未有自动化制备报道,也未有后续的临床试验。
技术实现要素:
[0005]
本发明制备了手性纯的l-抗坏血酸
18
f标记物 (4r, 5r)[
18
f]faa以及其他3个手性纯的旋光异构体,并在all-in-one自动化合成装置上完成f-18标记,标记产率30%左右(未经矫正)。临床前生物数据显示(4r, 5r)[
18
f]faa在荷瘤小鼠体内有一定的肿瘤摄取。临床研究数据表明:(4r, 5r)[
18
f]faa在肾癌以及肠癌患者中肿瘤放射性摄取一般,但出乎意料的是,(4r, 5r)[
18
f]faa检测甲癌以及肺癌效果极佳,肿瘤原发灶以及转移灶摄取均非常高。
[0006]
本发明采用两步法标记合成手性纯的l-抗坏血酸类似物放射性化合物(4r, 5r)[
18
f]faa,使其能用于研究aa在人体内分布、肿瘤诊断以及辐射计量学。
[0007]
本发明提供的一种手性纯的l-抗坏血酸类似物放射性化合物(4r, 5r)[
18
f]faa,
其具有式(ⅰ)所示的结构式:本发明还提供了(4r, 5r)[
18
f]faa其他三种构型,如式(ⅲ)所示:本发明的再一方面提供了上述的手性纯的l-抗坏血酸类似物放射性化合物(4r, 5r)[
18
f]faa的制备方法,包括:1) 将式ⅱ所示化合物a溶于无水乙腈中,得到含前体的无水乙腈;2)将含前体的无水乙腈与
18
f的k2.2.2/k2co3固体混合后,于80℃下加热10min,110℃下以氮气吹干溶剂,然后,向反应瓶中加入四氢呋喃以及浓酸,110℃,加热反应10分钟,得到标记溶液;所述四氢呋喃:浓酸=1:1;所述浓酸为浓硫酸或浓盐酸;3)反应结束后,以冰水浴冷却至0℃,向所述标记溶液中加入1n的氢氧化钠溶液,直至ph值为3-4之间,或与 pbs混合,得到混合溶液;4)将所述混合溶液利用氧化铝固相萃取柱和/或半制备柱子分离即得;所述分离时以ph 值 3.6、50 mm醋酸钠溶液为流动相,流速为3ml/min。
[0008]
在根据本发明的一个实施方案中,制备过程是通过手动或基于allinone模块自动化完成的。
[0009]
在根据本发明的一个实施方案中,步骤1)中以mg:ml计,化合物a:无水乙腈=20:3。
[0010]
在根据本发明的一个实施方案中,所述
18
f的k2.2.2/k2co3固体是经过qma固相萃取小柱吸附
18
f后,再以淋洗液淋所述qma固相萃取小柱收集得到
18
f的k2.2.2/k2co3溶液,再经加热、氮气流、无水乙腈共沸蒸干后得到的;所述淋洗液是将10mg k2.2.2和1.8mg k2co3溶于0.84 ml无水乙腈及0.16ml水中,配制得到的;以mg:mg:ml:ml计,所述k2.2.2:k2co3:无水乙腈:水=250:45:21:4。
[0011]
在根据本发明的一个实施方案中,还包括得到(4r, 5r)[18f]faa后在带放射性检测器的高效液相色谱(radio-hplc)中测定纯度。
[0012]
在根据本发明的一个实施方案中,所述测定纯度包括:第一流动相为0.1%三氟乙酸的水溶液,第二流动相为乙腈溶液,梯度洗脱条件:0~15 min,95%的第一流动相+5%第二流动相;流动相的流速为1 ml/min。
[0013]
本发明进一步提供了一种用于诊断或检测用途的组合物,其包含上述的 [
18
f]faa化合物。
[0014]
本发明进一步提供了上述的 [
18
f]faa化合物在制备用于追踪l-抗坏血酸在人体内分布、肿瘤诊断或辐射计量学中的试剂或药物中的应用。
[0015]
本发明的另一方面提供了上述手性纯aa类似物放射性化合物(4r, 5r)[
18
f]faa,在研究aa在人体的分布、肿瘤的诊断及辐射计量学等方面的应用。
[0016]
本发明的上述技术方案的有益效果如下:本发明提供的放射性探针(4r, 5r)[
18
f]faa,是aa的类似物,是手性纯化合物,可用于研究aa在人体内分布的研究、肿瘤的诊断、辐射计量学等方面。
附图说明
[0017]
图1为根据本发明实施例2实施案例2的自动化合成装置结构示意图。
[0018]
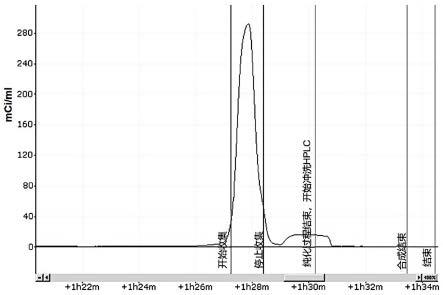
图2为根据本发明实施例2制备的半制备产品的hplc图谱。
[0019]
图3为根据本发明实施例2制备的合成产品(4r, 5r)[
18
f]faa的hplc图谱。
[0020]
图4为根据本发明实施例2、3的(4r, 5r)[
19
f]faa 的hplc图谱。
[0021]
图5为根据本发明实施例3的(4r, 5r)[
19
f]faa的合成路线图。
[0022]
图6为本发明实施例4中荷瘤鼠瘤内注射动态显像及延迟显像显像图。
[0023]
图7为本发明实施例4中荷瘤鼠尾静脉注射静态显像图。
[0024]
图8为本发明实施例5中不同肿瘤患者的pet/ct显像图。
[0025]
图9为本发明实施例5中患者甲癌术后全身多发转移的动态pet/ct显像图。
[0026]
图10为本发明实施例6中主要器官的平均时间-活性曲线。
[0027]
图11为本发明实施例7中主要器官的(4r, 5r)[
18
f]faa停留时间表。
[0028]
图12为本发明实施例7中主要器官的辐射剂量估算表。
[0029]
附图标记说明1-18
ꢀꢀ
第一-十八三通阀
ꢀꢀꢀꢀꢀꢀꢀꢀ
19-25
ꢀꢀꢀ
第一-七电磁阀26
ꢀꢀꢀꢀꢀ
三通电磁阀
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
ꢀ27
ꢀꢀ
18
f传输管开关28
ꢀꢀꢀꢀꢀ
18
o水收集瓶
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
29
ꢀꢀꢀꢀꢀꢀ
带加热功能的反应器30
ꢀꢀꢀꢀꢀ
无菌产品瓶
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
ꢀ31
ꢀꢀꢀ
废液瓶32
ꢀꢀꢀꢀꢀ
无菌滤膜
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
ꢀ33
ꢀꢀ
通气滤膜
34
ꢀꢀꢀꢀꢀ
qma固相萃取小柱
ꢀꢀꢀꢀ
ꢀ35
ꢀꢀꢀ
辅助加载试剂瓶试剂的注射器36
ꢀꢀꢀꢀꢀ
氧化铝固相萃取小柱
ꢀꢀꢀꢀ
37
ꢀꢀꢀ
hplc系统38
ꢀꢀꢀꢀꢀ
加载18f离子的注射筒
ꢀꢀꢀꢀ
39
ꢀꢀꢀ
hplc中的半制备柱40
ꢀꢀꢀꢀꢀ
hplc中的六通阀a 加载k
2.2.2
/k2co3溶液的注射器 b
ꢀꢀꢀꢀ
装载缓冲溶液的缓冲瓶c 装载前体的试剂瓶
ꢀꢀꢀꢀꢀꢀꢀꢀ
ꢀd
ꢀꢀꢀ
装载四氢呋喃和浓盐酸的试剂瓶e
ꢀꢀꢀ
装载vitc的试剂瓶
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀfꢀꢀꢀ
水袋
具体实施方式
为使本发明要解决的技术问题、技术方案和优点更加清楚,下面将结合附图及具体实施例进行详细描述。
[0030]
术语和定义acn,anhydrous acetonitrile,无水乙腈pbs,phosphate buffer saline,磷酸缓冲盐溶液反应式实施例1:(4r, 5r)[
18
f]faa的手动制备将2毫克前体即式(ⅱ)中所示的化合物a溶于0.3毫升acn中;将10mg k2.2.2和1.8mg k2co3溶于0.84 ml acn及0.16ml水中,配成淋洗液;99mg 醋酸钠、1.429克醋酸溶于500 ml水中获得ph 值为3.6的50 mm醋酸钠溶液;用10ml 1m nahco3及10ml ddh2o激活 qma柱;将
18
f-溶液过qma,再以淋洗液淋qma,收集洗脱液,110℃下以氮气吹干溶剂,加入1 ml无水乙腈,并于相同条件下除去溶剂,重复2次,含前体的无水乙腈加入标记反应瓶内,80℃下加热10min,110℃下以氮气吹干溶剂,向反应瓶中加入0.3毫升四氢呋喃以及0.3毫升浓硫酸,110℃,加热10分钟,冰水浴冷却至0℃,向标记溶液中加入1n的氢氧化钠溶液,直至ph值为3-4之间,混合溶液利用半制备柱子分离,流动相即为50 mm醋酸钠溶液,ph 值 3.6。
[0031]
实施例2 :(4r, 5r)[
18
f]faa的自动化制备在如图1所示的allinone模块上合成(4r, 5r)[
18
f]faa,由加速器生产的含放射性
18
f-离子通过氮气传到qma固相萃取小柱34上,由加载k
2.2.2
/k2co3溶液的注射器(a)注入k2.2.2/k2co3溶液将
18
f-从qma 固相萃取小柱34上淋到带加热功能的反应器29中,在开放条件下加热蒸发除去水分。
[0032]
通过装载前体的试剂瓶(c)将0.6 ml 含有3mg环硫酸酯化合物1的乙腈溶液加入带加热功能的反应器29中,加热到110℃反应10min。
[0033]
开放条件下蒸发除去乙腈,加入0.6 ml四氢呋喃和浓盐酸(v:v=1:1)在110℃反应10min。通过装载缓冲溶液的缓冲瓶(b)向带加热功能的反应器29中加入pbs,将混合液过氧化铝固相萃取小柱36后传输至hplc中的半制备柱39上分离,分离柱为c18柱,流动相为乙酸钠和冰乙酸的混合溶液,流速3ml/min,半制备效果图见图2,最高的放射性峰为目标产物,收集此峰的液体至无菌产品瓶30,即可获得高纯度、高比活度的产物。
[0034]
通过装载vitc的试剂瓶e向纯化产物中加入vitc抗氧化,过无菌滤膜32即可得产品。制备用时为40min,合成效率为30%。合成产品的hplc结果见图3,保留时间为9.317min,纯度》95%。(4r, 5r)[
18
f]faa的冷化合物(4r, 5r)[
19
f]faa的hplc结果分析见图4,其保留时间为9.206min。产品与(4r, 5r)[
19
f]faa在hplc上的保留时间基本一致,证明合成产品确实为(4r, 5r)[
18
f]faa。
[0035]
实施例3 :(4r, 5r)[
19
f]faa的合成合成线路图如图5所示。化合物2:将化合物1(5 g,17 mmol)溶于50 ml无水二甲基甲酰胺,向混合溶液中依次加入碘甲烷(4.8 g,34 mmol)以及碳酸钾(3.5 mg,25.5 mmol)。室温下反应过夜后,加入300 ml乙酸乙酯,用水(50 ml
ꢀ×ꢀ
2)以及饱和食盐水(50 ml)洗涤。有机相用无水硫酸镁干燥,过滤除去固体杂质。利用旋转蒸发仪减压除去滤液中的有机相,用乙酸乙酯/石油醚(v/v,2/8)过硅胶柱分离,收集目标组分,减压除去溶剂,得到4 g淡黄色油状物(产率:82%)。
[0036]
化合物3:将化合物2(2 g,6.9 mmol)溶于20 ml无水二甲基甲酰胺,向混合溶液中加入醋酸铜(20 mg,2 mmol)。加热回流30分钟。冷却至室温后,利用旋转蒸发仪减压除去溶剂,用乙酸乙酯/石油醚(v/v,7/3)过硅胶柱分离,收集目标组分,减压除去溶剂,得到1.1 g淡黄色油状物(产率:64%)。
[0037]
化合物4:将化合物3(500 mg,2 mmol)溶于10 ml吡啶中,向混合液中加入 对甲基苯磺酰氯 (380 mg, 2 mmol),室温下搅拌2小时。旋转蒸发仪减压除去有机相,用乙酸乙酯/石油醚(v/v,3/7)过硅胶柱分离,收集目标组分,减压除去溶剂,得到506 mg淡黄色油状物(产率:63%)。
[0038]
化合物5:将化合物4(500 mg,1.24 mmol)溶于10 ml无水二氯甲烷,向混合溶液中依次加入3,4-二氢吡喃酮(210 mg,2.5 mmol)以及吡啶对甲苯磺酸盐(62 mg,0.25 mmol)。室温下反应过夜后,利用旋转蒸发仪减压除去有机相,用乙酸乙酯/石油醚(v/v,2/8)过硅胶柱分离,收集目标组分,减压除去溶剂,得到337 mg淡黄色油状物(产率:56%)。
[0039]
化合物6:将化合物5(100 mg,0.21 mmol)溶于5 ml四氢呋喃中,向混合溶液中加入四丁基氟化铵的四氢呋喃溶液(1m,0.5 ml)室温下搅拌过夜。随后加入5毫升40%的硫酸,加热回流2小时。冷却至室温后,利用乙酸乙酯萃取(10 ml
ꢀ×ꢀ
3),合并有机相,利用旋转蒸发仪减压除去有机溶剂,用二氯甲烷/甲醇/醋酸(v/v/v,6/1/1)过硅胶柱分离,收集目标组分,减压除去溶剂,得到12 mg淡黄色油状物(产率:31%)。
[0040]
实施例4 :(4r, 5r)[
18
f]faa用于不同注射方式荷瘤小鼠micro-pet/ct动态显像建立hct8异种移植瘤裸鼠模型,麻醉荷瘤鼠后瘤内注射后进行动态显像:1ml注射器针取0.1 ml(约100μci)加/不加过饱和抗坏血酸钠的用生理盐水稀释自动化制备所得的溶液,用自动注射泵给模型裸鼠予瘤内注射0.1ml/min,行动态1h micropet/ct显像。进行延迟显像:注射2h后行静态显像。
[0041]
麻醉荷瘤鼠后尾静脉注射后1小时后行micro-pet/ct静态显像。荷瘤鼠瘤内注射动态显像及延迟显像显像图如图6所示。荷瘤鼠尾静脉注射静态显像图如图7所示。
[0042]
瘤内注射动态显像中显像剂(4r, 5r)[
18
f]faa在肿瘤中聚集,经过肿瘤组织吸收进入全身循环,经肝脏、肾脏排泄,加过饱和抗坏血酸钠能明显抑制肝脏中(4r, 5r)[
18
f]faa的代谢,注射后5min加/不加过饱和抗坏血酸钠肿瘤的suvmax(%id/g)分别为165、88。经瘤内加/不加过饱和抗坏血酸钠、尾静脉注射(4r, 5r)[
18
f]faa 1h后显像小鼠肿瘤的suvmax(%id/g)分别为45、34、6.3。不加过饱和抗坏血酸钠瘤内注射(4r, 5r)[
18
f]faa后2h延迟显像小鼠肿瘤的suvmax(%id/g)为35。(4r, 5r)[
18
f]faa在肿瘤中表现出高摄取,是良好的肿瘤示踪剂。抗坏血酸钠能显著抑制(4r, 5r)[
18
f]faa在肝脏中的代谢,但只能部分抑制肿瘤的摄取,说明瘤内注射(4r, 5r)[
18
f]faa是经过弥散作用和转运体的转运进入肿瘤细胞的,瘤内注射的方式或能通过弥散作用增加aa到达肿瘤细胞中的剂量和滞留时间。
[0043] 实施例5:(4r, 5r)[
18
f]faa用于肿瘤患者pet/ct动态显像6名患有不同肿瘤的患者在接受pet/ct扫描之前禁食至少6小时并排尿,在注射(4r, 5r)[
18
f]faa(平均448
±
113 mbq)后约5、13、30、45和60分钟,使用umi 780扫描仪(united imaging healthcare)或discovery mi扫描仪(ge healthcore)对每位参与者进行五次连续全身动态pet/ct扫描。umi 780扫描仪的ct采集参数为120 kv、智能100-200 mas、探测器覆盖范围40mm、间距0.9875、切片厚度1.0mm和旋转时间0.5s。pet发射扫描时间为每个床位1.5分钟,6个床位覆盖扫描范围。最后,通过正则化有序子集期望最大化算法(r-osem,united imaging)重建pet图像,并使用acct图像进行衰减校正。discovery mi扫描仪的ct采集参数为120 kv、智能120-180ma、探测器覆盖范围40mm、切片厚度0.625 mm、节距和速度0.984:1、39.37和旋转时间0.6s。pet发射扫描时间为每个床位1分钟,6至8个床位覆盖扫描范围。最后,通过贝叶斯惩罚似然(bpl)重建算法(q.clear,ge healthcare)重建pet图像,并使用acct图像进行衰减校正。在最后一次pet/ct扫描后收集尿样,并评估尿液总放射性。
[0044]
不同肿瘤患者的pet/ct显像图如图8所示。患者甲癌术后全身多发转移的动态pet/ct显像图如图9所示。肿瘤与背景的良好对比导致了良好的肿瘤成像,除6例肝转移外,6例患者的(4r, 5r)[
18
f]faa检测到2例原发性病灶和168例转移。所有肿瘤病变中(4r, 5r)[
18
f]faa的平均suvmax为6.89
±
3.82(范围1.62-22.85,中位数6.07)。肿瘤病变中(4r, 5r)[
18
f]faa的放射性摄取随时间快速而稳定地增加。(4r, 5r)[
18
f]faa在人体内使用是安全的,在肿瘤中表现出高摄取。
[0045]
实施例6:人体内生物分布对于6组通过实施案例5得到的图像,在第一个时间点的全身图像中描绘roi,并复制到随后的时间点扫描中,以确定每个源器官中的累积放射性活度,必要时手动调整生殖器官体积。主要来源器官包括大脑、甲状腺、肺、心壁、肝脏、胆囊、胃、肾上腺、肾脏、胰腺、脾脏、子宫(仅限女性)、睾丸(仅限男性)和骨髓。生物分布分析包括肌肉,吸收剂量分析包括膀胱。脊椎roi被绘制为红骨髓的替代物。从每个roi导出时间-活性曲线,每个源器官的累积放射性表示为注射剂量的百分比(%id)。
[0046]
主要器官的平均时间-活性曲线如图10所示。结果显示所有受试者表现出相似的生物分布特征。在最早的成像时间点,大多数受试者在肝脏、肾上腺、肾脏、脉络丛、垂体和
甲状腺中都有[18f]dfa的高摄取。除肾脏外,这些器官在注射(4r, 5r)[
18
f]faa后1小时仍表现出高摄取。(4r, 5r)[
18
f]faa在脾脏、肠道、唾液腺和骨髓中表现出中等摄取,随着时间的推移,示踪剂清除率缓慢。示踪剂主要通过肾脏排泄到膀胱,随着时间的推移,膀胱中的活性积累逐渐增加。在成像阶段,心脏、肌肉和大脑的摄取量一直很低。大多数器官的活性呈指数下降,可以用单指数或双指数函数拟合。(4r, 5r)[
18
f]faa在人体中显示出与aa相似的生物分布模式,并在肿瘤中以适当的动力学表现出高摄取和滞留。
[0047]
实施例7:辐射计量学对于6组通过实施案例6得到的数据,通过单指数或双指数函数模型的曲线拟合,将停留时间计算为每个时间-活性曲线下的面积。膀胱的停留时间是通过从时间零点到最后一次发射扫描时间的时间-活动曲线的积分加上从最后一次扫描时间到无穷大(仅物理衰减)的函数的积分而产生的。随后,这些停留时间用于辐射吸收剂量计算。使用医用内部辐射剂量(mird)算法计算每个受试者的辐射吸收剂量。根据标准化cristy eckerman 70 kg成年男性模型计算mird指定目标区域的吸收剂量,并根据国际放射防护委员会出版物中的组织加权因子评估有效剂量。
[0048]
主要器官的(4r, 5r)[
18
f]faa停留时间表如图11所示,辐射剂量估算表如图12所示。结果显示停留时间最长的器官是肝脏,为22.5
±
4.94e
−
02小时,然后是肺部(3.18
±
0.49 e
−
02小时),脾脏(2.19
±
1.59e
−
02小时),肾脏(1.77
±
0.53e
−
03 h)和膀胱壁(1.16
±
0.54e
−
03小时)。胸腺的停留时间最低(2.74
±
1.37e
−
05 h),全身停留时间为9.87
±
2.75e。吸收剂量最高的器官是肝脏(3.42
±
0.75e
−
02 mgy/mbq),脾脏(2.95
±
1.70e
−
02 mgy/mbq)、肾上腺(1.89
±
0.61e
−
02 mgy/mbq)和肾脏(1.87
±
0.38e
−
02mgy/mbq),最低的是脑(4.88
±
1.21e
−
03mgy/mbq)。平均全身吸收剂量为6.54
±
1.58e-03 msv/mbq。平均有效剂量(ed)为1.68
±
0.36 e
−
02 msv/mbq。(4r, 5r)[
18
f]faa的全身吸收有效剂量与最广泛使用的pet示踪剂[18f]fdg(1.90
±
0.36 e-02 msv/mbq)相似。
[0049]
以上所述是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明所述原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1