一种全氟-2,3-环氧-2-甲基戊烷的制备方法及其应用与流程
1.本发明属于氟精细化学合成领域,具体涉及一种全氟-2,3-环氧-2-甲基戊烷的制备方法及其应用。
背景技术:
2.哈龙(溴氟烷烃类)类灭火剂由于化学结构稳定,沸点低,在紫外线的作用下可释放游离的溴原子破坏臭氧层,已逐渐被市场淘汰。替代哈龙和氟代烷类的环境友好的高效灭火剂中比较突出的产品是美国3m公司研制的商品名为novec 1230
tm
防火液体,俗称全氟己酮,其化学式为cf3cf2cocf(cf3)2。全氟己酮是在常温下为液体,易于保存和运输;易于气化,稍微采取技术措施即可转移热量像气体灭火剂那样灭火抑爆。因此,全氟己酮作为一种洁净灭火剂,突出优点在于灭火高效、安全、环保和日常维护方便。
3.国内外报道的全氟己酮合成路线中,南京理工大学液态合成路线可获得纯度高达90%以上的全氟己酮粗产物,其合成路线如下:(a)以六氟丙烯为原料并添加催化剂,采用气相齐聚法或液相齐聚法将六氟丙烯进行齐聚反应,制备得到全氟-4-甲基-2-戊烯(d1)、少量全氟-2-甲基-2-戊烯(d2)、三聚体。其中,二聚体和三聚体的比例受溶剂影响最大,可通过改变溶剂及溶剂的配比选择性生成所需产物。(b)在极性非质子溶剂中,以碱金属氟化物为催化剂,无水条件下,可将d1转变为d2。(c)以d2为原料,采用氧化剂将d2氧化为全氟-2,3-环氧-2-甲基-戊烷。(d)在溶剂和催化剂条件下,将d2环氧化物全氟-2,3-环氧-2-甲基-戊烷中间体异构化,得到全氟-2-甲基-3-戊酮(全氟己酮)。
4.在放大生产过程中,上述方法4个步骤中步骤(c)得到的产物纯度、收率最低;氧化剂次氯酸钠溶液的有效氯只有8%-14%,而且随着反应的进行,有效氯含量降低,需要延长反应时间使d2完全环氧化,因此用量较大且反应较慢。其中,d2环氧化物纯度较低,是因为有约10%的d2并未发生反应;同时由于d2的沸点与全氟己酮沸点非常接近,反应结束后通过一次精馏纯化,全氟己酮纯度只能达到97%-98%,很难达到要求的99%以上,因此需要重复氧化、重复精馏、提高精馏塔高度,但这将增大设备投资成本和延长工艺路线,加大了后期纯化难度。此外,上述反应过程还会产生大量次氯酸钠废水,并且次氯酸钠腐蚀不锈钢。
5.进一步地,d2环氧化物全氟-2,3-环氧-2-甲基-戊烷是一种重要的有机合成中间体,可用于合成含氟醇类化合物,亦可与其他亲电试剂反应,从而使其分子结构内含有氟原子。含氟类化合物由于其自身的生物活性和人体内分子伪拟性而受到极大关注,使得全氟-2,3-环氧-2-甲基戊烷具有应用潜力。而且全氟-2,3-环氧-2-甲基戊烷的制备方法主要是通过全氟-2-甲基-2-戊烯氧化而得。但其他合成方法,如三乙胺氧化物氧化法,该方法是使用三乙胺氧化物作为氧化剂,n,n
’‑
二甲基甲酰胺为溶剂,在-30℃下反应1小时,收率98%。该方法氧化效率高,反应时间短,但氧化物制备较为困难,且危险较大,同时反应温度较低,放大生产时成本较高。因此,优化全氟-2,3-环氧-2-甲基-戊烷的制备方法具有一定的现实意义。
技术实现要素:
6.针对现有技术中存在的问题和不足,本发明的目的在于提供一种全氟-2,3-环氧-2-甲基戊烷的制备方法及其应用。
7.基于上述目的,本发明采用如下技术方案:
8.本发明第一方面提供了一种全氟-2,3-环氧-2-甲基戊烷的制备方法,所述制备方法为:在0-20℃的温度条件下,将相转移催化剂、有机卤代氧化剂和水加入反应釜中,然后再向反应釜中加入全氟-2-甲基-2-戊烯进行反应,反应结束后过滤收集滤液,滤液静置分液后收集有机相,得到全氟-2,3-环氧-2-甲基戊烷。
9.优选地,所述有机卤代氧化剂为卤代异氰尿酸类化合物。
10.优选地,所述有机卤代氧化剂为卤代异氰尿酸或/和卤代异氰尿酸盐。
11.优选地,所述卤代异氰尿酸为三氯异氰尿酸、二氯异氰尿酸、二溴异氰尿酸中的一种或几种;所述卤代异氰尿酸盐为二氯异氰尿酸钠、二溴异氰尿酸钠的一种或几种。
12.更加优选地,所述卤代异氰尿酸为三氯异氰尿酸。
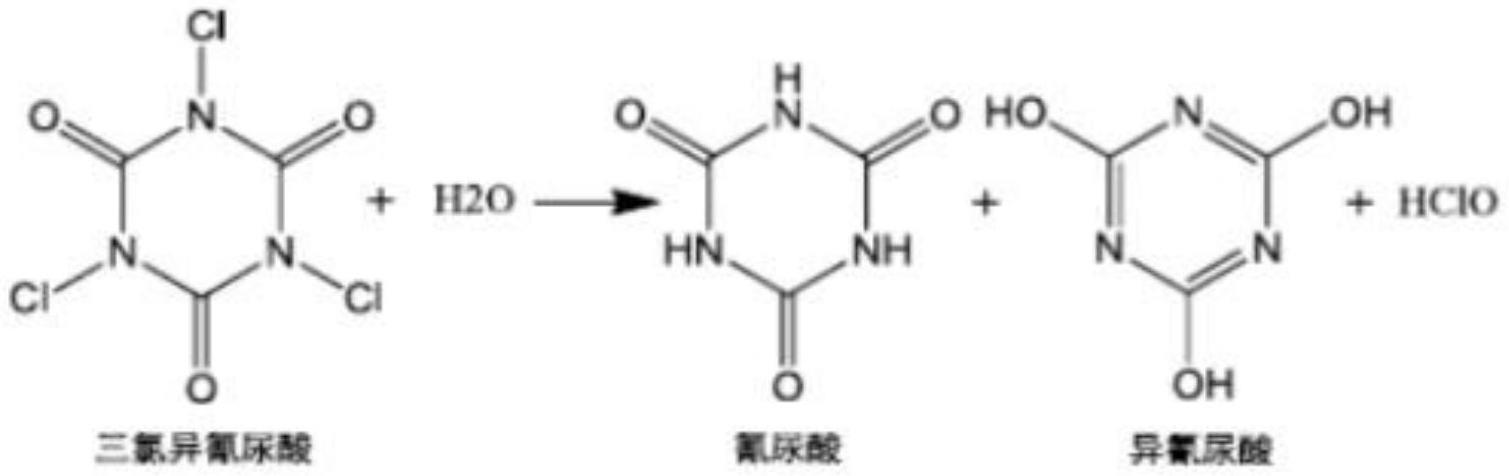
13.进一步地,三氯异氰尿酸与水可发生水解反应释放有效氯,具体反应如下所示:
[0014][0015]
优选地,所述水的摩尔质量是有机卤代氧化剂中卤素摩尔质量的1.1-5倍。更加优选地,所述水与有机卤代氧化剂中卤素摩尔质量的1.1-3.5倍。
[0016]
更加优选地,所述水和三氯异氰尿酸的摩尔比为(3.3-15)∶1。
[0017]
优选地,所述相转移催化剂为乙腈、四乙二醇二甲醚、四丁基溴化铵、吡啶中的一种或几种。
[0018]
优选地,所述相转移催化剂与水的摩尔比为(0.05-0.25)∶1。更加优选地,所述相转移催化剂与水的摩尔比为(0.05-0.15)∶1。
[0019]
更加优选地,所述乙腈与水的摩尔比为(0.05-0.15)∶1;更加优选地,乙腈与水的摩尔比为0.1∶1。
[0020]
优选地,所述有机卤代氧化剂中卤素摩尔质量是全氟-2-甲基-2-戊烯摩尔质量的1.1-3倍。
[0021]
更加优选地,所述三氯异氰尿酸与全氟-2-甲基-2-戊烯的摩尔比为(0.37-1)∶1。
[0022]
进一步地,三氯异氰尿酸和水反应生成的次氯酸作为氧化剂与全氟-2-甲基-2-戊烯发生亲核环氧化反应,反应过程如下所示:
[0023][0024]
进一步地,反应结束后得到的反应混合物中,过量的次卤酸以及乙腈在水相中;产物全氟-2,3-环氧-2-甲基戊烷和少量的原料d2在有机相中;副产物氰尿酸、异氰尿酸不溶于水,可溶于碱性溶液,因此可通过过滤和分离水相以去除。
[0025]
更加优选地,收集所述有机相后还包括后处理过程,所述后处理过程为精馏。
[0026]
优选地,向反应釜中加入全氟-2-甲基-2-戊烯之前,将水加入反应釜中时需要向反应釜中加入碳酸盐或/和碳酸氢盐,以促进卤代异氰尿酸类化合物更快地形成次氯酸,释放有效氯。
[0027]
优选地,所述碳酸盐与水的摩尔比为(0.1-0.3)∶1;所述碳酸氢盐与水的摩尔比为(0.2-0.6)∶1。更加优选地,所述碳酸盐与水的摩尔比为(0.1-0.2)∶1;所述碳酸氢盐与水的摩尔比为(0.2-0.4)∶1。
[0028]
优选地,反应过程中收集反应釜中产生的气体,并对收集的气体进行冷凝处理,回收气体中的全氟-2-甲基-2-戊烯和全氟-2,3-环氧-2-甲基戊烷。更加优选地,当在反应釜中加入所述碳酸盐或/和碳酸氢盐时,副产物二氧化碳会带走大量产物和原料,因此反应过程中还需加装冷凝回流装置,用以回收被二氧化碳带走的全氟-2-甲基-2-戊烯和全氟-2,3-环氧-2-甲基戊烷。
[0029]
优选地,所述反应时间为2-8h。更加优选地,当反应釜中还加入所述碳酸盐或碳酸氢盐时,所述反应时间为2-4h。
[0030]
本发明第二方面提供了一种全氟己酮的制备方法,包括如下步骤:
[0031]
(1)六氟丙烯直接合成全氟-2-甲基-2-戊烯:以极性非质子溶剂为介质,碱金属氟化物、相转移催化剂为催化剂,在无水条件,20-30℃温度下,六氟丙烯进行反应,得到全氟-4-甲基-2-戊烯;反应结束后,升温至50-80℃继续以全氟-4-甲基-2-戊烯为原料进行异构化反应,得到全氟-2-甲基-2-戊烯;
[0032]
(2)制备全氟-2,3-环氧-2-甲基戊烷:以步骤(1)得到的全氟-2-甲基-2-戊烯为原料,按上述第一方面任一所述制备方法制备全氟-2,3-环氧-2-甲基戊烷;
[0033]
(3)制备全氟己酮:以极性非质子溶剂为介质,碱金属氟化物为催化剂,在无水条件,50-65℃温度下,以步骤(2)得到的全氟-2,3-环氧-2-甲基戊烷为原料进行反应,得到全氟己酮。
[0034]
更加优选地,步骤(1)中所述极性非质子溶剂为乙腈,相转移催化剂为18-冠-6,碱金属氟化物为氟化铯(csf)和氟化钾(kf)的混合物或氟化铯(csf)。
[0035]
更加优选地,步骤(1)中所述极性非质子溶剂、相转移催化剂、碱金属氧化物、六氟丙烯四者摩尔比为(3-5)∶1∶1∶(1-20)。
[0036]
更加优选地,步骤(3)中所述极性非质子溶剂为乙腈,碱金属氟化物为氟化铯(csf)。
[0037]
更加优选地,步骤(3)中所述极性非质子溶剂、碱金属氟化物、全氟-2,3-环氧-2-甲基戊烷三者摩尔比为(3-5)∶1∶(1-10)。
[0038]
更加优选地,步骤(1)中所述异构化反应时间为6-12h;步骤(3)中所述反应时间为4-8h。
[0039]
更加优选地,步骤(3)中反应结束后可对反应液进行后处理,所述后处理过程为一次精馏。
[0040]
与现有技术相比,本发明的有益效果如下:
[0041]
(1)本技术首次采用有机氯化合物如三氯异氰尿酸作为d2环氧化的氧化剂,利用其与水可发生水解反应释放大量次氯酸,进而氧化d2发生环氧化反应,具有产物纯度、收率高,反应速率快,耗时短,加入体积小,可提高容器使用效率等优点。这是因为,卤代异氰尿酸或卤代异氰尿酸盐如三氯异氰尿酸有效氯含量高;而且其自身呈粉末或粒状固体,能在水中稳定释放有效氯,并将其持续稳定地提供给后续环氧化反应;反应结束后生成的副产物氰脲酸、异氰尿酸基本无毒,无二次污染;且其自身不溶有机物,易于分离,减少后处理纯化难度。因此,卤代异氰尿酸或卤代异氰尿酸盐可有效提高产物的转化纯度及收率。如在其中一项实施例中,本技术在扩大生产至300g d2的情况下,本技术制备的d2环氧化物产物收率达97.0%,纯度达96.8%;而采用常用次氯酸钠作氧化剂的产品收率和纯度均已跌至90%上下。
[0042]
(2)本发明还在卤代异氰尿酸或卤代异氰尿酸盐与水反应过程中加入碳酸盐或碳酸氢盐,促使卤代异氰尿酸或卤代异氰尿酸盐持续释放并保持一定的有效氯浓度,不仅能有效调节有效氯浓度,还能加快反应时间。但其反应生成的副产物二氧化碳气体会带走一部分原料及产物,因此加装冷凝回流装置后还能进一步提高反应转化效率及产品收率。
[0043]
(3)本发明还采用一步法合成d2,结合本发明制备的d2环氧化物进行全氟己酮的制备,仅经简易精馏即可得到纯度≥99%的全氟己酮。因此,本发明全氟-2,3-环氧-2-甲基戊烷制备方法具有实际有效的应用价值。
[0044]
(4)本技术采用的卤代异氰尿酸或卤代异氰尿酸盐还有贮运稳定,成型和使用方便,对不锈钢几乎无腐蚀作用等优点。
具体实施方式
[0045]
为使本发明的目的、技术方案及优点更加清楚明白,以下通过实施例对本发明作进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
[0046]
需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本技术。
[0047]
实施例1:全氟-2,3-环氧-2-甲基戊烷的制备方法中碳酸氢钠的加入及冷凝处理的探讨
[0048]
为了探讨碳酸氢钠的加入及冷凝处理对反应时长和产物全氟-2,3-环氧-2-甲基戊烷的纯度和收率的影响,发明人分别做了以下实验,即实施例1-1至实施例1-3和对比例1,其对应的碳酸氢钠的加入及冷凝处理如表1所示,相应的反应时长和产物全氟-2,3-环氧-2-甲基戊烷的纯度和收率结果如表1所示。
[0049]
实施例1-1
[0050]
本发明实施例提供一种全氟-2,3-环氧-2-甲基戊烷的制备方法,包括如下步骤:
[0051]
(1)开启制冷装置,将14.4ml乙腈(0.28mol)、50g水(2.8mol)加入烧瓶中搅拌均匀,再向烧瓶中加入85g三氯异氰尿酸(0.37mol);在烧瓶上部加装恒压漏斗,并向恒压漏斗中加入300g全氟-2-甲基-2-戊烯(d2,1mol),备用。
[0052]
(2)待温度稳定在0℃左右后开启烧瓶内部的搅拌装置,打开恒压漏斗开关,将d2缓慢滴加进入烧瓶,滴加完毕后,继续搅拌8h进行环氧化反应,反应结束后得到反应混合物。
[0053]
(3)将步骤(2)得到的反应混合物过滤除去副产物异氰尿酸与氰尿酸,收集滤液静置分液,收集下层有机相,即得产物301.5g全氟-2,3-环氧-2-甲基戊烷。计算产物收率为95.4%,纯度96.2%。
[0054]
实施例1-2
[0055]
本发明实施例提供一种全氟-2,3-环氧-2-甲基戊烷的制备方法,包括如下步骤:
[0056]
(1)开启制冷装置,将14.4ml乙腈(0.28mol)、50g水(2.8mol)、62.2g碳酸氢钠(0.74mol)加入烧瓶中搅拌均匀,再向烧瓶中加入85g三氯异氰尿酸(0.37mol);在烧瓶上部加装恒压漏斗,并向恒压漏斗中加入300g全氟-2-甲基-2-戊烯(d2,1mol),备用。
[0057]
(2)待温度稳定在0℃左右后开启烧瓶内部的搅拌装置,打开恒压漏斗开关,将d2缓慢滴加进入烧瓶,滴加完毕后,继续搅拌3h进行环氧化反应,反应结束后得到反应混合物。
[0058]
(3)将步骤(2)得到的反应混合物过滤除去副产物异氰尿酸与氰尿酸,收集滤液静置分液,收集下层有机相,即得产物281.8g全氟-2,3-环氧-2-甲基戊烷。计算产物收率为89.2%,纯度96.5%。
[0059]
实施例1-3
[0060]
本发明实施例提供一种全氟-2,3-环氧-2-甲基戊烷的制备方法,包括如下步骤:
[0061]
(1)开启制冷装置,将14.4ml乙腈(0.28mol)、50g水(2.8mol)、62.2g碳酸氢钠(0.74mol)加入烧瓶中搅拌均匀,再向烧瓶中加入85g三氯异氰尿酸(0.37mol);在烧瓶上部加装恒压漏斗,并向恒压漏斗中加入300g全氟-2-甲基-2-戊烯(d2,1mol),备用;同时在恒压漏斗上部加装冷凝管,冷凝温度为-5℃。
[0062]
(2)待温度稳定在0℃左右后开启烧瓶内部的搅拌装置,打开恒压漏斗开关,将d2缓慢滴加进入烧瓶,滴加完毕后,继续搅拌3h进行环氧化反应,反应结束后得到反应混合物。
[0063]
(3)将步骤(2)得到的反应混合物过滤除去副产物异氰尿酸与氰尿酸,收集滤液静置分液,收集下层有机相,即得产物306.5g全氟-2,3-环氧-2-甲基戊烷。计算产物收率为97.0%,纯度96.8%。
[0064]
对比例1
[0065]
本发明对比例提供一种全氟-2,3-环氧-2-甲基戊烷的制备方法,包括如下步骤:
[0066]
(1)开启制冷装置,将200ml乙腈(3.8mol)、1700g次氯酸钠溶液(有效氯10%,次氯酸根摩尔量为2.28mol)加入烧瓶;在烧瓶上部加装恒压漏斗,并向恒压漏斗中加入300g全氟-2-甲基-2-戊烯(d2,1mol),备用。
[0067]
(2)待温度稳定在0℃左右后开启烧瓶内部的搅拌装置,打开恒压漏斗开关,将d2缓慢滴加进入烧瓶,滴加完毕后,继续搅拌15h进行环氧化反应,反应结束后得到反应混合物。
[0068]
(3)将步骤(2)得到的反应混合物静置分液,收集下层有机相,即得产物285g全氟-2,3-环氧-2-甲基戊烷。计算产物收率为90.2%,纯度89.1%。
[0069]
表1本发明实施例和对比例的参数
[0070][0071]
由表1可知,本发明实施例1-1仅加三氯异氰尿酸制备的产物d2环氧化物收率为95.4%,纯度96.2%,比对比例1次氯酸钠制备的产物收率高出5.2%,纯度高出7.1%,反应时长缩短近1倍。这是因为,三氯异氰尿酸有效氯含量可高达90%,而次氯酸钠有效氯仅10%左右,在反应过程中能为d2的环氧化反应提供高浓度的有效氯,有利于d2充分进行环氧化,进而缩短反应时长。因此,卤代异氰尿酸类化合物三氯异氰尿酸的加入可有效提高产物收率、纯度,缩短反应时长。
[0072]
进一步地,对比实施例1-1至1-3可以看出,在氧化剂水解反应过程中添加碳酸氢钠(实施例1-2)虽然可以有效缩短反应时长,但其收率降至89.2%,纯度96.5%;而在反应过程中添加冷凝处理后(实施例1-3),产物收率高达97.0%,纯度96.8%。这是因为,碳酸氢钠虽然可以促进次氯酸根离子的释放,进一步调节有效氯浓度,加快反应速率,缩短反应时间,但其副产物二氧化碳的溢出会带走部分原料和产物,所以加装冷凝回流装置回收原料和产物后,得到的产物收率有了明显改善。因此,在氧化剂水解反应过程中添加碳酸氢钠时,建议增设冷凝回流装置以提高产物收率及纯度。
[0073]
此外,由于氧化剂三氯异氰尿酸本身呈固体,加入体积小,反应后生成的副产物也不溶于有机相,经过滤和分液即可有效滤除,后处理操作简单方便;同时由于氧化剂有效氯含量高,d2充分进行环氧化,进一步增加产物纯度。
[0074]
实施例2:全氟-2,3-环氧-2-甲基戊烷的制备方法中不加碳酸氢钠时原料加入量的探讨
[0075]
为了探讨不加碳酸氢钠时原料加入量对反应时长和产物全氟-2,3-环氧-2-甲基戊烷的纯度和收率的影响,发明人分别做了以下实验,即实施例1-1、实施例2-1至2-2和对比例1至3,其对应的原料d2加入量和氧化剂种类如表2所示,相应的反应时长和产物全氟-2,3-环氧-2-甲基戊烷的纯度和收率结果如表2所示。
[0076]
实施例2-1
[0077]
一种全氟-2,3-环氧-2-甲基戊烷的制备方法与实施例1-1的内容基本相同,其不同之处在于:步骤(1)中乙腈的加入量为4ml(0.078mol),水的加入量为13.7g(0.76mol),三氯异氰尿酸的加入量为30.2g(0.13mol),d2的加入量为100g(0.33mol);步骤(2)中环氧化反应时长为8h;步骤(3)中得到99.3g全氟-2,3-环氧-2-甲基戊烷,产物收率为94.3%,纯度96.5%。
[0078]
实施例2-2
[0079]
一种全氟-2,3-环氧-2-甲基戊烷的制备方法与实施例1-1的内容基本相同,其不同之处在于:步骤(1)中乙腈的加入量为7ml(0.14mol),水的加入量为25g(1.39mol),三氯异氰尿酸的加入量为44.1g(0.19mol),d2的加入量为150g(0.5mol);步骤(2)中环氧化反应时长为8h;步骤(3)中得到151.7g全氟-2,3-环氧-2-甲基戊烷,产物收率为96.0%,纯度95.9%。
[0080]
对比例2
[0081]
一种全氟-2,3-环氧-2-甲基戊烷的制备方法与对比例1的内容基本相同,其不同之处在于:步骤(1)中乙腈的加入量为70ml(1.33mol),次氯酸钠溶液的加入量为567g(次氯酸根摩尔量为0.76mol),d2的加入量为100g(0.33mol);步骤(2)中环氧化反应时长为12h;步骤(3)中得到94.6g全氟-2,3-环氧-2-甲基戊烷,产物收率为89.8%,纯度90.2%。
[0082]
对比例3
[0083]
一种全氟-2,3-环氧-2-甲基戊烷的制备方法与对比例1的内容基本相同,其不同之处在于:步骤(1)中乙腈的加入量为120ml(2.34mol),次氯酸钠溶液的加入量为818g(1.1mol),d2的加入量为150g(0.5mol);步骤(2)中环氧化反应时长为12h;步骤(3)中得到141.4g全氟-2,3-环氧-2-甲基戊烷,产物收率为89.5%,纯度90.8%。
[0084]
表2本发明实施例和对比例的参数
[0085][0086]
由表2可知,将d2加入量相同的实施例和对比例比较后发现,如对比例3和实施例2-2,反应时长相差达到4h,产物收率相差6.5%,纯度相差5.1%。这是因为,三氯异氰尿酸的有效氯含量高达90%,可以提供充足的hclo使d2充分被氧化,产物中只存在微量的d2,因此产物纯度有明显的提高。因此,本发明氧化剂三氯异氰尿酸相比常用次氯酸钠结果均具明显优势。
[0087]
对比实施例可以看出,随着原料d2加入量的增加,环氧化反应时长保持不变,产物收率呈现略有上升的趋势,产物纯度基本不变。这是因为,随着原料投入量的增加,生产的扩大,原料及产物的挥发带来的损失减少,因此,本发明在d2添加量为300g时,产物收率和纯度优势更加明显。
[0088]
进一步地,将对比例1-3对比后可以发现,将原料d2由100g扩大生产至300g后,次氯酸钠溶液已达到1700g。可以预见地,进一步扩大生产后,次氯酸钠废液进一步增加,这给产物后处理及废液处理都带来极大地困难。
[0089]
实施例3:全氟-2,3-环氧-2-甲基戊烷的制备方法中加碳酸氢钠不冷凝处理时原料加入量的探讨
[0090]
为了探讨加碳酸氢钠但不进行冷凝回流时原料加入量对反应时长和产物全氟-2,3-环氧-2-甲基戊烷的纯度和收率的影响,发明人分别做了以下实验,即实施例1-2、实施例3-1至3-2和对比例1至3,其对应的原料d2加入量和氧化剂种类如表3所示,相应的反应时长和产物全氟-2,3-环氧-2-甲基戊烷的纯度和收率结果如表3所示。
[0091]
实施例3-1
[0092]
一种全氟-2,3-环氧-2-甲基戊烷的制备方法与实施例1-2的内容基本相同,其不同之处在于:步骤(1)中乙腈的加入量为4ml(0.078mol),水的加入量为13.7g(0.76mol),碳酸氢钠的加入量为16.8g(0.2mol),三氯异氰尿酸的加入量为30.2g(0.13mol),d2的加入量为100g(0.33mol);步骤(2)中环氧化反应时长为3h;步骤(3)中得到94.5g全氟-2,3-环氧-2-甲基戊烷,产物收率为89.7%,纯度96.2%。
[0093]
实施例3-2
[0094]
一种全氟-2,3-环氧-2-甲基戊烷的制备方法与实施例1-2的内容基本相同,其不同之处在于:步骤(1)中乙腈的加入量为7ml(0.14mol),水的加入量为25g(1.39mol),碳酸氢钠的加入量为28.6g(0.34mol),三氯异氰尿酸的加入量为44.1g(0.19mol),d2的加入量为150g(0.5mol);步骤(2)中环氧化反应时长为3h;步骤(3)中得到142.5g全氟-2,3-环氧-2-甲基戊烷,产物收率为90.2%,纯度96.8%。
[0095]
表3本发明实施例和对比例的参数
[0096][0097]
由表3可知,将d2加入量相同的实施例和对比例比较后发现,如对比例3和实施例3-2,反应时长相差达到9h,产物收率相差0.7%,纯度相差6%。这是因为,碳酸氢钠虽可加快反应速率,缩短反应时间,但其副产物二氧化碳的溢出会带走部分原料和产物,导致产物
收率下降。因此,当体系中添加碳酸氢钠但不进行冷凝处理时,本发明三氯异氰尿酸相比常用次氯酸钠结果收率相近但纯度优势明显。
[0098]
实施例4:全氟-2,3-环氧-2-甲基戊烷的制备方法中加碳酸氢钠且进行冷凝处理时原料加入量的探讨
[0099]
为了探讨加碳酸氢钠且进行冷凝回流时原料加入量对反应时长和产物全氟-2,3-环氧-2-甲基戊烷的纯度和收率的影响,发明人分别做了以下实验,即实施例1-3、实施例4-1至4-2和对比例1至3,其对应的原料d2加入量和氧化剂种类如表4所示,相应的反应时长和产物全氟-2,3-环氧-2-甲基戊烷的纯度和收率结果如表4所示。
[0100]
实施例4-1
[0101]
一种全氟-2,3-环氧-2-甲基戊烷的制备方法与实施例1-3的内容基本相同,其不同之处在于:步骤(1)中乙腈的加入量为4ml(0.078mol),水的加入量为13.7g(0.76mol),碳酸氢钠的加入量为16.8g(0.2mol),三氯异氰尿酸的加入量为30.2g(0.13mol),d2的加入量为100g(0.33mol);步骤(2)中环氧化反应时长为3h;步骤(3)中得到100.3g全氟-2,3-环氧-2-甲基戊烷,产物收率为95.2%,纯度96.5%。
[0102]
实施例4-2
[0103]
一种全氟-2,3-环氧-2-甲基戊烷的制备方法与实施例1-3的内容基本相同,其不同之处在于:步骤(1)中乙腈的加入量为7ml(0.14mol),水的加入量为25g(1.39mol),碳酸氢钠的加入量为28.6g(0.34mol),三氯异氰尿酸的加入量为44.1g(0.19mol),d2的加入量为150g(0.5mol);步骤(2)中环氧化反应时长为3h;步骤(3)中得到152.2g全氟-2,3-环氧-2-甲基戊烷,产物收率为96.3%,纯度97.2%。
[0104]
表4本发明实施例和对比例的参数
[0105][0106]
由表4可知,将d2加入量相同的实施例和对比例比较后发现,如对比例3和实施例4-2,反应时长相差达到9h,产物收率相差6.8%,纯度相差6.4%。这是因为,冷凝装置将原料和产物大部分回收,损失量很少,收率得以提高。因此,当体系中添加碳酸氢钠且进行冷凝处理时,本发明氧化剂三氯异氰尿酸相比常用次氯酸钠结果均具明显优势。
[0107]
实施例5:全氟己酮制备方法的优化
[0108]
为了探讨六氟丙烯合成d2的制备方法以及全氟-2,3-环氧-2-甲基戊烷的制备方法对反应时长和产物全氟-2,3-环氧-2-甲基戊烷的纯度和收率的影响,发明人分别做了以下实验,即实施例5-1、实施例5-2和对比例4-5,其对应的六氟丙烯合成d2的制备方法和全
氟-2,3-环氧-2-甲基戊烷的制备方法如表5所示,相应的反应时长和产物全氟己酮的纯度和收率结果如表5所示。
[0109]
实施例5-1
[0110]
本发明实施例提供一种全氟己酮的制备方法,包括如下步骤:
[0111]
(1)六氟丙烯直接合成d2:
[0112]
a)将400ml溶剂乙腈(7.8mol)、合成d1的催化剂8g csf(0.053mol)、合成d2的催化剂17.4g kf(0.3mol)、79.2g 18-冠-6(0.3mol)加入5l的反应釜中,开启加热和搅拌。
[0113]
b)待温度稳定在25℃左右时,开始通入六氟丙烯气体,至压力达到0.5mpa左右停止进料,反应一段时间后压力下降,然后按压力进料,待压力下降至0.3mpa继续进料;3000g的六氟丙烯(20mol)完成进料,待压力下降至0后,合成全氟-4-甲基-2-戊烯(d1)的反应结束,反应耗时4h左右。
[0114]
c)加热反应釜温度至70℃左右,持续搅拌8h进行反应;反应结束后将反应釜降温至0-5℃,将反应液静置分液后收集下层有机相,即得2868g产物全氟-2-甲基-2-戊烯(d2),计算产物收率为95.6%,纯度92.5%。
[0115]
(2)d2环氧化生成d2环氧化物:
[0116]
d)开启制冷装置,将14.4ml乙腈(0.28mol)、50g水(2.8mol)、62.2g碳酸氢钠(0.74mol)加入烧瓶中搅拌均匀,再向烧瓶中加入85g三氯异氰尿酸(0.37mol);在烧瓶上部加装恒压漏斗,并向恒压漏斗中加入300g全氟-2-甲基-2-戊烯(d2,1mol),备用;同时在恒压漏斗上部加装冷凝管,冷凝温度为-5℃。
[0117]
e)待温度稳定在0℃左右后开启烧瓶内部的搅拌装置,打开恒压漏斗开关,将d2缓慢滴加进入烧瓶,滴加完毕后,继续搅拌3h进行环氧化反应,反应结束后得到反应混合物。
[0118]
f)将反应混合物过滤除去副产物异氰尿酸,收集滤液静置分液,收集下层有机相,即得产物295g全氟-2,3-环氧-2-甲基戊烷。
[0119]
(3)d2环氧化物异构化生成全氟己酮:
[0120]
g)在无水条件下,将200ml溶剂乙腈(3.8mol)、15.2g催化剂csf(0.1mol)、300g全氟-2,3-环氧-2-甲基戊烷(0.949mol)加入烧瓶中,烧瓶上部加装球型回流管,开启加热和搅拌。
[0121]
h)待温度稳定在55℃左右后,回流反应12h。反应结束后,冷却降温后收集反应液,将反应液静置分液收集下层有机相,有机相进行一次精馏处理,得到282.9g全氟己酮。计算产物的综合收率为84.2%,纯度99.5%。
[0122]
实施例5-2
[0123]
(1)六氟丙烯合成d1:
[0124]
a)将200ml溶剂乙腈(3.8mol)、合成d1的催化剂8g csf(0.053mol)加入5l的反应釜中,开启加热和搅拌。
[0125]
b)待温度稳定在25℃左右时,开始通入六氟丙烯气体,至压力达到0.5mpa左右停止进料,反应一段时间后压力下降,然后按压力进料,待压力下降至0.3mpa开始进料;3000g的六氟丙烯(20mol)完成进料后,待压力下降至0后,合成全氟-4-甲基-2-戊烯(d1)的反应结束,反应耗时4h左右。将反应釜降温至0-5℃后倒出反应液,静置分液后收集下层有机相,即得2886g产物d1,计算产物收率为96.2%,纯度94.2%。
[0126]
(2)d1异构化生成d2:
[0127]
c)将200ml溶剂乙腈(3.8mol)、合成d2的催化剂17.4gkf(0.3mol)、79.2g 18-冠-6(0.3mol)、2886g d1(9.62mol)加入反应釜中,开启加热和搅拌。
[0128]
d)待温度稳定在70℃左右时,持续搅拌8h进行反应;反应结束后将反应釜降温至0-5℃,将反应液静置分液后收集下层有机相,即得2747g产物全氟-2-甲基-2-戊烯(d2),计算产物收率为95.2%,纯度92.2%。
[0129]
(3)d2环氧化生成d2环氧化物:
[0130]
e)开启制冷装置,将14.4ml乙腈(0.28mol)、50g水(2.8mol)、62.2g碳酸氢钠(0.74mol)加入烧瓶中搅拌均匀,再向烧瓶中加入85g三氯异氰尿酸(0.37mol);在烧瓶上部加装恒压漏斗,并向恒压漏斗中加入300g全氟-2-甲基-2-戊烯(d2,1mol),备用;同时在恒压漏斗上部加装冷凝管,冷凝温度为-5℃。
[0131]
f)待温度稳定在0℃左右后开启烧瓶内部的搅拌装置,打开恒压漏斗开关,将d2缓慢滴加进入烧瓶,滴加完毕后,继续搅拌3h进行环氧化反应,反应结束后得到反应混合物。
[0132]
g)将反应混合物过滤除去副产物异氰尿酸,收集滤液静置分液,收集下层有机相,即得产物295g全氟-2,3-环氧-2-甲基戊烷。
[0133]
(4)d2环氧化物异构化生成全氟己酮:
[0134]
h)在无水条件下,将200ml溶剂乙腈(3.8mol)、15.2g催化剂csf(0.1mol)、300g全氟-2,3-环氧-2-甲基戊烷(0.949mol)加入烧瓶中,烧瓶上部加装球型回流管,开启加热和搅拌。
[0135]
i)待温度稳定在55℃左右后,回流反应12h。反应结束后,冷却降温后收集反应液,将反应液静置分液收集下层有机相,有机相进行一次精馏处理,得到282g全氟己酮。计算产物的综合收率为80.7%,纯度99.2%。
[0136]
对比例4
[0137]
一种全氟己酮的制备方法与实施例5-1的内容基本相同,其不同之处在于:
[0138]
步骤d)中乙腈的加入量为200ml(3.8mol),次氯酸钠溶液的加入量为1700g(次氯酸根摩尔量为2.28mol),d2的加入量为300g(1mol);步骤e)中环氧化反应时长为15h;步骤f)中得到267.6g全氟-2,3-环氧-2-甲基戊烷,产物收率为84.7%,纯度87.1%。
[0139]
步骤h)中得到240g全氟己酮。计算产物的综合收率为73.0%,纯度97.8%。
[0140]
对比例5
[0141]
一种全氟己酮的制备方法与实施例5-2的内容基本相同,其不同之处在于:
[0142]
步骤e)乙腈的加入量为200ml(3.8mol),次氯酸钠溶液的加入量为1700g(次氯酸根摩尔量为2.28mol),d2的加入量为300g(1mol);步骤e)中环氧化反应时长为15h;步骤f)中得到267.6g全氟-2,3-环氧-2-甲基戊烷,产物收率为84.7%,纯度87.1%。
[0143]
步骤i)中得到240.8g全氟己酮。计算产物的综合收率为70.0%,纯度98.0%。
[0144]
表5本发明实施例和对比例的参数
[0145][0146]
由表5可以看出,对比实施例5-1和实施例5-2,1步法收率更大。这是因为,d1会在静置分液的过程挥发,而1步法去掉d1的后处理过程,减少了d1的损失,所以产物收率变大;但1步法合成d2会需要反应釜同时具有升降温功能,会提高设备成本。因此,可综合考虑生产成本和产品收率选择合适的d2合成过程。
[0147]
同时比较实施例5-1与对比例4、实施例5-2与对比例5,可以看出,以1步法制备全氟己酮,本发明氧化剂参与的制备方法优势更大,总时长减少9h,收率提高10%左右,纯度可经一次精馏后就可达到99%。因此,优选采用一步法合成d2,并以本发明氧化剂参与的制备方法制备全氟己酮。
[0148]
综上所述,本发明有效克服了现有技术中的不足,且具高度产业利用价值。上述实施例的作用在于说明本发明的实质性内容,但并不以此限定本发明的保护范围。本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1