一种基于改性POSS互穿网络协同增强增韧的环氧树脂及其制备方法
一种基于改性poss互穿网络协同增强增韧的环氧树脂及其制备方法
技术领域
1.本发明涉及环氧树脂技术领域,具体涉及一种基于改性poss互穿网络协同增强增韧的环氧树脂及其制备方法。
背景技术:
2.环氧树脂由于其优异的力学性能和热性能,被广泛用作高性能纤维增强复合材料的基体。此外,它是最通用的商业热固性树脂,用于各种场景,如粘合剂、涂料和结构部件基体等。但是由于纯环氧树脂固化后呈三维交联网络结构,存在脆性大、抗冲击性差等缺点。因此,环氧树脂的增韧一直是相关领域的关键研究课题之一。
3.目前关于环氧树脂的基本增韧改性方法包括:1)通过物理共混引入第二相;2)通过加入柔性主链等方式对环氧树脂进行化学改性,3)构建互穿网络结构(ipn)。然而,单一的韧性提高往往伴随着其他性能的下降。
4.比如cn106146857a公开了端羧基液体氟橡胶增韧改性环氧树脂体系的制备方法,所得橡胶增韧体系的断裂伸长率最高可提升45%,但橡胶增韧剂的使用同时也会导致体系玻璃化温度和模量的明显下降。综合而言,构建ipn结构的增韧方法可以整合各个组分的优点,有望全面提高体系性能。cn114085491a公开了一种聚醚醚酮改性环氧树脂组合物的制备方法,通过将各组原料按照配方比例混合对普通环氧树脂进行改性,显著改善了其柔韧性能。但由于相形态的控制困难和有限的处理灵活性,在进一步应用方面存在一定挑战。
5.考虑到单一增韧方法的局限性,已有研究人员尝试将两种或多种单一增韧剂结合起来,以寻求环氧树脂性能的全面改善。然而,单一增韧剂的简单组合通常难以提高环氧树脂的性能,有时甚至会导致力学性能降低。例如,marouf(marouf,b.t.;pearson,r.a.;bagheri,r.anomalous fracture behavior in an epoxy-based hybrid composite[j].mat.sci.eng.a-struct.2009,515(1-2):49-58.)将纳米粘土和核壳橡胶颗粒添加到环氧树脂基体中,发现与单组分增韧方法相比,复合增韧体系的断裂韧性显著降低,他们认为这是由于不同增韧组分之间的机制的竞争作用。
[0006]
多面体低聚倍半硅氧烷(poss)的直径为1-3nm,是二氧化硅的最小存在形式。它具有内部硅/氧核心以及外部有机取代基的特殊结构,前者可明显增强改性树脂基体的热机械性能和物理性能,后者则使其具有极大的可设计性。上述优点使其成为实现协同增韧的最有希望的候选材料之一。然而,与其他纳米填料一样,poss也饱受团聚问题的困扰,因此,未改性poss力学性能的改善在很大程度上受到限制。已经有很多关于poss改性的研究,以获得更好的分散性和更高的韧性,但同时增加了工艺的复杂性。因此,一种简单有效的、能同时提升环氧树脂强度和韧性的改性制备方法是目前高性能树脂及其复合材料研究领域亟待解决的问题。
技术实现要素:
[0007]
本发明针对环氧树脂增韧和强度无法兼得的问题,提供一种基于改性poss互穿网络协同增强增韧的环氧树脂,利用改性poss和乙烯基酯树脂(ver)形成的加聚网络和环氧树脂固化网络共同形成全互穿网络结构增韧体系,实现高效增韧的同时实现增强,获得具有高冲击性能和强度的环氧树脂。
[0008]
为实现上述目的,本发明采用的技术方案是:
[0009]
一种基于改性poss互穿网络协同增强增韧的环氧树脂,包括如下组分:环氧树脂、乙烯基酯树脂、改性poss、固化剂、引发剂和促进剂;
[0010]
所述改性poss为八乙烯基倍半硅氧烷(ovposs)与3-巯基丙酸衍生物反应得到。
[0011]
本发明通过设计一种具有特殊结构的改性poss同时实现反应性和相容性,其中未反应的乙烯基键可与乙烯基酯基体进行自由基聚合反应,而改性的杂原子基团一方面与基体具有结构相似性,另一方面可通过物理缠结与周围基体相容,最终实现纳米级分散。此外,本发明通过引入与环氧树脂分子结构高度相似的乙烯基酯树脂建立全互穿网络结构,实现了在非相分离的情况下的综合性能的全面提高(包括失效前的塑性形变能力)。在此“初步”增强增韧的基础上,改性poss首先通过化学交联和物理缠结整合入互穿网络进行增强,并在受到外力时通过颗粒脱粘触发周围基体的塑性剪切屈服从而大幅吸收能量,以实现韧性的显著提高。
[0012]
所述改性poss的合成过程包括步骤:将ovposs(乙烯基poss)和3-巯基丙酸衍生物在溶剂中混合,在自由基引发剂作用下反应,去除溶剂得到所述改性poss。本发明中以3-巯基丙酸衍生物与乙烯基poss反应,通过巯基-烯点击反应,以具有巯基的柔性链化合物取代ovposs八角笼上的部分乙烯基,使得所制备改性poss同时具有聚合反应性和基体相容性,因而能在整合入互穿网络结构的基础上充分发挥其特殊结构带来的性能优势。
[0013]
得到的eposs的结构如下式:
[0014][0015]
优选地,所述3-巯基丙酸衍生物包括3-巯基丙酸乙酯(emp)、3-巯基丙酸异辛酯(imp)、3-巯基丙酸十八烷酯(omp)、3-巯基丙酸(amp)中任一种。进一步优选的,所述3-巯基
丙酸衍生物为3-巯基丙酸乙酯(emp),随着链长的增加,物理缠结效应增加,基体的交联密度降低,而物理缠结可通过物理拓扑作用限制自由体积的增大,因而保证了室温下的机械性能(杨氏模量、拉伸强度、硬度等)。
[0016]
所述ovposs和3-巯基丙酸衍生物的摩尔比为1:3-5;优选地ovposs和3-巯基丙酸衍生物的摩尔比为1:4;其中3-巯基丙酸衍生物取代ovposs上任意位置的乙烯基,其分子结构如下式中任一种;
[0017][0018]
反应温度为70-90℃,反应时间为4-8h;
[0019]
所述溶剂包括甲苯、二甲苯、四氢呋喃中一种或多种;所述自由基引发剂包括aibn和/或bpo;自由基引发剂使用量为3-巯基丙酸衍生物的1-3wt%;反应在惰性气体保护下进行;惰性气体包括氮气、氩气中任一种。
[0020]
所述环氧树脂为双酚a型环氧树脂;优选e54、e51、e44、e42中的一种或多种;所述乙烯基酯树脂中苯乙烯溶剂体积含量为30-45%;若苯乙烯溶剂过少,则会导致乙烯基酯树脂乃至于增韧树脂体系粘度过大,一方面,这会使得在树脂浇筑时气泡不易排出,另一方面,还使得体系难以通过机械搅拌充分分散进而影响后续试样性能;若苯乙烯溶剂过多,则会导致所制备增韧体系交联密度过大,脆性增大,与增韧目的产生相反的效果。优选地,所述乙烯基酯树脂中苯乙烯溶剂体积含量为30-40%,进一步优选35-40%。
[0021]
所述固化剂包括甲基四氢苯酐(methpa)、四氢苯酐(thpa)、六氢苯酐(hhpa)中一种或多种;所述自由基聚合引发剂包括bpo和/或tbpo;所述促进剂包括dmp-30和/或dmp-10;
[0022]
所述环氧树脂和乙烯基酯树脂的质量比为0.8-1.2:0.8-1.2;当环氧树脂与乙烯基酯之间比例过高或过低时,会导致flory-huggins方程中混合自由能过大导致分相,而这
一方面会使得后续碳纤维复合材料制备时海岛相沿纤维分布,进而导致性能提高受限,另一方面,不可避免地引入的复杂的相调控工艺会使得生产成本增加。优选地,所述环氧树脂和乙烯基酯树脂的质量比为1:1;
[0023]
所述eposs与乙烯基酯树脂的质量比1-10:100;随着eposs的加入,拉伸强度和杨氏模量呈现上升趋势,冲击强度出现先上升后下降的趋势
[0024]
优选地,所述eposs与乙烯基酯树脂的质量比2-8:100,该比例下环氧树脂的综合性能更为优异。
[0025]
所述固化剂与环氧树脂的质量比为80-90:100;所述引发剂和乙烯基酯树脂的质量比为1-3:100,用于引发乙烯基酯聚合,所述促进剂与环氧树脂的质量比为0.5-2:100,促进环氧固化。
[0026]
本发明还提供所述的基于改性poss互穿网络协同增强增韧的环氧树脂的制备方法,包括步骤:环氧树脂水浴加热,加入乙烯基酯树脂和eposs搅拌混合,再加入固化剂搅拌混合,超声处理后加入引发剂和促进剂搅拌混合,固化得到所述环氧树脂。
[0027]
水浴加热温度为40-70℃;在该温度范围下,既能保证较低粘度,使得机械搅拌顺利进行,也不会出现单体间的提前反应。
[0028]
超声时温度为40-70℃,超声时间15-60min;超声后改性poss在体系内分散效果更好,进一步减少团聚的现象。
[0029]
每次搅拌混合的时间为10min以上;优选10-60min,或10-30min;
[0030]
固化程序为85-105℃/4h,130-150℃/3h,170-190℃/2h。
[0031]
与现有技术相比,本发明具有以下有益效果:
[0032]
(1)本发明合成的eposs兼具良好的反应性和相容性,其中未反应的乙烯基键可与乙烯基酯基体进行自由基聚合反应,而改性的杂原子基团一方面与基体具有结构相似性,另一方面可通过物理缠结与周围基体相容,最终实现纳米级分散。
[0033]
(2)本发明使用非相分离原位增韧技术,制备了以ver/eposs加聚网络和ep固化网络所构建的全互穿网络结构增韧体系,其中,整个体系没有出现相分离,避免了调控相形貌的复杂工艺过程。
[0034]
(3)本发明制备的增韧环氧树脂相对于未改性环氧树脂,在优选配比下(eposs的用量仅约为体系总质量的2wt%),拉伸强度提高了17.92%,杨氏模量提高了9.0%,冲击韧性的提升了139.6%。
[0035]
(4)通过廉价的原料与简单的方法实现了高效增强增韧,具有较大的实际可行性与工业化潜力。
附图说明
[0036]
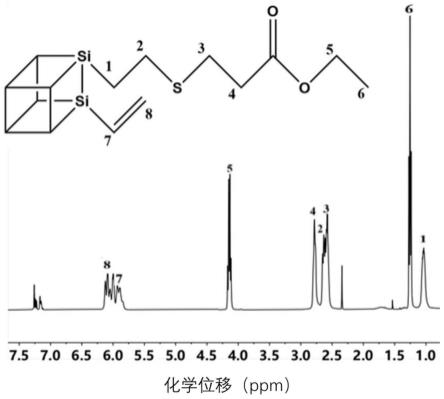
图1为实施例1合成的eposs的核磁氢谱图。
[0037]
图2为实施例1合成的eposs的核磁硅谱图
[0038]
图3为e/v/eposs-6试样的tem高分辨形貌图。
[0039]
图4为e/v/ovposs-6试样的tem形貌图。
[0040]
图5为e/v/ovposs-6试样的sem团聚形貌图。
[0041]
图6为e/v/ovposs-6试样在图8团聚形貌下的硅元素能谱mapping图。
[0042]
图7为纯ep的sem拉伸断口形貌图,左边为1k倍,右边为20k倍。
[0043]
图8为e/v试样的sem拉伸断口形貌图,左边为1k倍,右边为20k倍。
[0044]
图9为e/v/eposs-6试样的sem拉伸断口形貌图,左边为1k倍,右边为20k倍。
具体实施方式
[0045]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。本领域技术人员在理解本发明的技术方案基础上进行修改或等同替换,而未脱离本发明技术方案的精神和范围,均应涵盖在本发明的保护范围内。
[0046]
本发明通过设计一种具有特殊结构的改性poss,其结构的特殊性可同时实现反应性和相容性;另一方面,本发明通过引入与环氧树脂分子结构高度相似的乙烯基酯树脂建立全互穿网络结构,实现了在非相分离的情况下的综合性能的全面提高(包括失效前的形变能力)。在此“初步”增强增韧的基础上,改性poss首先通过化学交联和物理缠结整合入互穿网络进行增强,强键合力使得应力均一分布,而物理缠结作用会限制因交联密度下降而引起的自由体积的增大,因而保证了试样在室温下机械强度的提升。此外,在受到大载荷时,体系可通过颗粒脱粘触发周围基体的塑性剪切屈服从而大幅吸收能量,以实现韧性的显著提高。改性poss支链的物理缠结在室温下是有效的增强机制,而poss脱粘引发的基体塑性剪切屈服是增韧的主要原因,而这两者的优势在ver/ep/poss互穿网络结构中能得到充分发挥。
[0047]
本发明首先合成一种可同时实现与树脂良好反应性和相容性的改性poss,合成步骤包括:将摩尔比为1:3-5的ovposs和3-巯基丙酸衍生物加入到溶剂中,室温搅拌至充分溶解和分散;氮气鼓泡纯化后,向烧瓶中加入引发剂搅拌后,将反应体系加热持续反应。旋转蒸馏除去溶剂,真空干燥后得到浅黄色或乳白色粘性液体为改性poss。
[0048]
再通过构建以改性poss为核心的全互穿ipn符合体系实现环氧树脂的全互穿增强增韧,构建步骤包括:将ver与环氧树脂混合,并加入固化剂。体系通过机械搅拌、超声分散获得均匀分布体系,加入引发剂与促进剂,体系在真空下脱气除气泡,于模具中加热固化,得到高韧性高强度的环氧树脂。
[0049]
以下具体实施方式中所采用的原料均购于现有市场;所指的“份”,均为重量份。
[0050]
实施例1
[0051]
将2g(3.159mmol)乙烯基poss和1.696g(12.638mmol)emp添加到含有50ml甲苯的250ml三颈烧瓶中。将三颈烧瓶置于带有磁力搅拌器的恒温水浴中,并充分搅拌,鼓泡纯化30min后,加入17.0mg aibn,继续鼓泡并搅拌10min。随后将反应体系加热至80℃,持续5h。反应后,通过旋转蒸馏除去甲苯,真空干燥24h后得到浅黄色粘性液体,用eposs表示。
[0052]
实施例2
[0053]
将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂methpa为85份。两者混合后加入1份实施例1制备的eposs,在55℃机械混合30min,然后在超声中分散30min后,加入引发剂bpo与促进剂dmp-30,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为85℃/4h,140℃/3h,180℃/2h。试样命名为e/v/eposs-1。
[0054]
实施例3
[0055]
将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂methpa为85份。两者混合后,加入3份实施例1制备的eposs,在55℃机械混合30min,然后在超声中分散30min后,加入引发剂bpo与促进剂dmp-30,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为85℃/4h,140℃/3h,180℃/2h。试样命名为e/v/eposs-3。
[0056]
实施例4
[0057]
将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂methpa为85份。两者混合后,加入6份实施例1制备的eposs,在55℃机械混合30min,然后在超声中分散30min后,加入引发剂bpo与促进剂dmp-30,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为85℃/4h,140℃/3h,180℃/2h。试样命名为e/v/eposs-6。
[0058]
实施例5
[0059]
将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂methpa为85份。两者混合后,加入10份实施例1制备的eposs,在55℃机械混合30min,然后在超声中分散30min后,加入引发剂bpo与促进剂dmp-30,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为85℃/4h,140℃/3h,180℃/2h。试样命名为e/v/eposs-10。
[0060]
对比例1
[0061]
将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂methpa为85份。两者混合后,在55℃机械混合30min,然后在超声中分散30min后,加入引发剂bpo与促进剂dmp-30,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为85℃/4h,140℃/3h,180℃/2h。试样命名为e/v。
[0062]
对比例2
[0063]
将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂methpa为85份。两者混合后加入1份未经改性的ovposs,在55℃机械混合30min,然后在超声中分散60min后,加入引发剂bpo与促进剂dmp-30,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为85℃/4h,140℃/3h,180℃/2h。试样命名为e/v/ovposs-1。
[0064]
对比例3-5
[0065]
按照对比例2的工艺,调整未改性ovposs的加入量分别为3份、6份和10份,分别得到e/v/ovposs-3、e/v/ovposs-6、e/v/ovposs-10。
[0066]
实施例6
[0067]
将2g(3.159mmol)乙烯基poss和2.759g(12.638mmol)3-巯基丙酸异辛酯(imp)添加到含有50ml甲苯的250ml三颈烧瓶中。将三颈烧瓶置于带有磁力搅拌器的恒温水浴中,并充分搅拌,鼓泡纯化30min后,加入27.59mg aibn,继续鼓泡并搅拌10min。随后将反应体系加热至70℃,持续6h。反应后,通过旋转蒸馏除去甲苯,真空干燥24h后得到乳白色粘性液体,用iposs表示。
[0068]
将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂methpa为80
份。两者混合后加入6份iposs,在70℃机械混合30min,然后在超声中分散15min后,加入引发剂bpo与促进剂dmp-10,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为100℃/4h,150℃/3h,190℃/2h。试样命名为e/v/iposs-6。
[0069]
实施例7
[0070]
将2g(3.159mmol)乙烯基poss和4.532g(12.638mmol)3-巯基丙酸十八烷酯(omp)添加到含有50ml二甲苯的250ml三颈烧瓶中。将三颈烧瓶置于带有磁力搅拌器的恒温水浴中,并充分搅拌,鼓泡纯化30min后,加入45.32mg aibn,继续鼓泡并搅拌10min。随后将反应体系加热至90℃,持续4h。反应后,通过旋转蒸馏除去甲苯,真空干燥24h后得到乳白色粘性液体,用oposs表示。
[0071]
将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂thpa为80份。两者混合后加入1份oposs,在55℃机械混合30min,然后在超声中分散40min后,加入引发剂bpo与促进剂dmp-30,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为90℃/4h,150℃/3h,190℃/2h。试样命名为e/v/oposs-3。
[0072]
实施例8
[0073]
将2g(3.159mmol)乙烯基poss和1.341g(12.638mmol)3-巯基丙酸(amp)添加到含有50ml四氢呋喃的250ml三颈烧瓶中。将三颈烧瓶置于带有磁力搅拌器的恒温水浴中,并充分搅拌,鼓泡纯化30min后,加入13.41mg bpo,继续鼓泡并搅拌10min。随后将反应体系加热至80℃,持续5h。反应后,通过旋转蒸馏除去甲苯,真空干燥24h后得到乳白色粘性液体,用aposs表示。
[0074]
将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂hhpa为90份。两者混合后加入1份aposs,在55℃机械混合30min,然后在超声中分散30min后,加入引发剂bpo与促进剂dmp-10,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为90℃/4h,130℃/3h,170℃/2h。试样命名为e/v/amposs-1。
[0075]
实施例9
[0076]
取苯乙烯溶剂体积分数为30%的乙烯基酯树脂(ver-1),将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂methpa为85份。两者混合后加入3份实施例1制备的eposs,在70℃机械混合30min,然后在超声中分散30min后,加入引发剂bpo与促进剂dmp-30,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为85℃/4h,130℃/3h,170℃/2h。试样命名为e/v-1/eposs-6。
[0077]
实施例10
[0078]
取苯乙烯溶剂体积分数为45%的乙烯基酯树脂(ver-2),将100份ver与100份环氧树脂体系混合,在环氧树脂体系中,固化剂methpa为85份。两者混合后加入3份实施例1制备的eposs,在55℃机械混合30min,然后在超声中分散30min后,加入引发剂bpo与促进剂dmp-30,质量比例为0.5:1。搅拌3min,体系在真空下脱气除气泡,然后倒入不锈钢模具中,在设定程序下加热固化,固化制度为100℃/4h,150℃/3h,190℃/2h。试样命名为e/v-2/eposs-6。
[0079]
性能测试
[0080]
1、数据表征
[0081]
将实施例1制备的eposs进行核磁扫描。
[0082]
如图1所示,将每个1h nmr信号的分配标记如下:6.27-5.68ppm(m,3h,8和7)、4.14ppm(q,2h,5)、3.01-2.70ppm(m、2h,4)、2.70-2.40ppm(m、4h、2和3)、1.25ppm(t,3h、6)、1.04ppm(m,2h,1)。上述定量结果表明,当ovposs:emp的化学计量比等于1:4时,ovposs上的一半乙烯基与emp反应。
[0083]
如图2所测的
29
si nmr谱所示,ovposs由于硅氧八角笼中硅原子的单一化学环境,仅在-79.67ppm处显示一个化学位移峰,而eposs在-68.76ppm处的另一个峰证明emp基团已成功与ovposs分子反应。此外,在eposs硅谱中的每个信号峰的放大图中有四个子峰(分别放大显示在左侧和右侧)。对于约-80.49ppm的r
1-si峰,包括四个连续的子峰,分别位于-80.68ppm、-80.56ppm、-80.42ppm和-80.30ppm,分别对应于(r
1-si)(r
1-si)3,(r
1-si)(r
1-si)2(r
2-si),(r
1-si)(r
1-si)(r
2-si)2,(r
1-si)(r
2-si)3,如图2所示。类似地,对于r
2-si峰,在-68.75ppm附近也有四个峰,对应于r2-si的四种化学情况:(r
2-si)(r
2-si)3(-68.68ppm),(r
2-si)(r
2-si)2(r
1-si)(-68.77ppm),(r
2-si)(r
2-si)(r
1-si)2(-68.84ppm),(r
2-si)(r
1-si)3(-68.94ppm)。
[0084]
综上,成功合成了具有四个乙烯基和四个emp基团的eposs。
[0085]
2、eposs的分散效果
[0086]
由于纳米粒子的分散性是决定其能否发挥机械性能优势的重要因素,因此对改性前后的试样的纳米粒子分散情况进行了表征。
[0087]
将实施例4制备的e/v/eposs-6和对比例4制备的e/v/ovposs-6进行tem扫描,观察其在基体中分散情况,结果分别如图3和图4,可见改性后的eposs在体系中分散均匀,呈现纳米级的分散形貌,无团聚现象;而图4中未改性的ovposs在体系中呈现明显团聚形貌,团聚畴区大小约为几微米。
[0088]
在图5sem图与图6硅元素能谱mapping扫描中,未改性的ovposs在基体中呈现明显的团聚形貌,这也极大地影响了其试样的力学性能(见表1)。
[0089]
3、力学性能
[0090]
对纯环氧树脂(ep)和实施例2-10、对比例1-5制备的环氧树脂进行力学性能测试。纯环氧树脂(ep)指只有环氧,无ver和引发剂,且固化剂和促进剂相同的试样。拉伸试验按gb t2567-2008标准进行测试,冲击试验按gbt 1043.1-2008标准进行测试,结果如表1所示。
[0091]
表1实施例和对比例的环氧树脂力学性能
[0092]
[0093][0094]
从表1可见,随着eposs的加入,机械性能呈现明显的增强。其中,拉伸强度不断增加,在e/v/eposs-6时达到最大值103.37mpa,相比于纯ep提升了17.92%。杨氏模量先略降低后再升高,最终可达2110mpa,比于纯ep提升了9.0%。断裂伸长率在ver加入时增大,这是由于互穿网络结构的构建对链段局域迁移能力的提升,而eposs的加入使其略有下降。对于冲击强度而言,eposs的加入使其大幅提升,其中e/v/eposs-6试样具有最大值37.88kj/m2,相比于纯ep提升了139.6%,后续则因为eposs出现团聚而出现下降趋势。
[0095]
机械性能的整体提升归因于以改性poss为增韧机制触发位点的全互穿网络结构。在承载时,改性poss首先通过未改性的乙烯基进行化学交联以及改性的支链与周围基体链段产生物理缠结整合入互穿网络进行增强。良好的键合使得应力均一分布,而物理缠结作用会限制因交联密度下降而引起的自由体积的增大,因而保证了试样在室温下机械强度的提升。此外,在受到大载荷时,体系可通过颗粒脱粘触发周围基体的塑性剪切屈服从而大幅吸收能量,以实现韧性的显著提高。改性poss支链的物理缠结在室温下是有效的增强机制,而poss脱粘引发的基体塑性剪切屈服是增韧的主要原因,而这两者的优势在ver/ep/poss互穿网络结构中能得到充分发挥。
[0096]
对于其他改性试样(e/v/iposs、e/v/oposs、e/v/amposs),可看到加入同样会使得机械性能呈现增强趋势,但力学性能表现不够理想,而eposs改性试样比同比例的其他试样综合力学性能更优越。
[0097]
实施例9和10的e/v-1/eposs-6、e/v-2/eposs-6,对于不同苯乙烯含量的树脂增韧体系,可看到其增韧效果弱于实施例5(e/v/eposs-6),事实上,若苯乙烯溶剂减少,则会导致乙烯基酯树脂乃至于增韧树脂体系粘度过大,这可能会使得在树脂浇筑时气泡不易排出,或使得体系难以通过机械搅拌充分分散进而影响后续试样性能;若苯乙烯溶剂过多,则会导致所制备增韧体系交联密度过大,脆性增大,与增韧目的产生相反的效果,如ver中苯乙烯溶剂进一步减少或增大,对结果是不利的。
[0098]
将纯样ep、对比例1的e/v和实施例4的e/v/eposs-6式样的拉伸断裂样品进行断口形貌观察,结果分别如图7-9。图7可见纯样ep中呈现明显的脆性断裂特征,而对比例1(图8)中e/v呈现明显的基体剪切屈服增韧机制的形貌,说明ver的引入形成的互穿网络结构一方面对树脂体系进行了整体的增韧与增强,另一方面也增大了基体的延展性,这使得基体在受到外界应力载荷时可通过塑性剪切变形来耗散能量;从图9中可见对于引入eposs的体系,e/v/eposs-6试样在塑性剪切变形形貌之外,还在更小尺度下呈现出纳米颗粒脱粘,而
poss脱粘后形成的空穴成为基体塑性剪切变形提供了位点与空间。在前者的作用下,后者作为主增韧机制被广泛触发,进而大幅提升了基体的韧性。图9圈出部分为“颗粒脱粘-基体剪切屈服”增韧机制的形貌,可看出其在试样中广泛分布。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1