合成5-己烯-1-醇的方法与流程
合成5-己烯-1-醇的方法
技术领域
1.本发明涉及5-己烯-1-醇配置技术领域,具体为合成5-己烯-1-醇的方法。
背景技术:
2.烯醇是一类重要的精细化学品,如异植醇、叶醇等,这些烯醇可进一步作为关键中间体用于合成具有高经济附加值的医药、农药、香精和香料等(chinese journal of catalysis,2021,42,2105-2121),烯醇有着较大的市场需求量。发明一种环保、经济,可以实现工业化生产的烯醇方法有着重要的意义。
3.5-己烯-1-醇是常见的有机合成中间体和医药中间体,也是化工、医药的重要原料。合成5-己烯-1-醇的方案主要有:方案一:(angewandte chemie-international edition,2019,vol.58,#4,p.1129-1133)用5-己烯酸甲酯或者5-己烯酸还原合成5-己烯-1-醇;方案二:(us4288642,1981)用1,6-己二醇合成得到5-己烯-1-醇;方案三:(wo2022/19921,2022)用2-(吲哚甲基)四氢吡喃合成得到5-己烯-1-醇;方案四:(cn114249629)用5-己炔-1-醇加氢反应得到5-己烯-1-醇。
4.但是现有合成5-己烯-1-醇的方法都有一定的缺点。方案一:用5-己烯酸甲酯或者5-己烯酸还原合成5-己烯-1-醇,不仅原料不容易获得,还原文献报道收率都不高,副产物也多,纯化困难。
5.方案二:该方法反应操作需要300度高温消除,反应温度高,且收率低。
6.方案三:该方法用2-(吲哚甲基)四氢吡喃作为原料,此原料昂贵且不易得到。
7.方案四:该方法用到pdzn/meso_s-c催化剂,使pd活性位点中毒,以降低催化剂活性为代价,提高烯醇产物的选择性。但这种催化剂不仅牺牲了活性金属pd的利用率,而且毒化剂pb、喹啉等会对环境产生污染,且催化剂的稳定性也较差。
8.为此,我们提出了合成5-己烯-1-醇的方法。
技术实现要素:
9.针对现有技术的不足,本发明提供了合成5-己烯-1-醇的方法,解决了上述背景技术中提出的问题。
10.为实现以上目的,本发明通过以下技术方案予以实现:合成5-己烯-1-醇的方法,其特征在于:包括以下步骤:
11.s1、以6-溴-1-己烯为原料,四丁基溴化铵作为催化剂,在乙腈溶剂中,加热与醋酸钾反应,反应完全;
12.s2、将混合液冷却至室温(18-25℃),反应液减压浓缩,浓缩至不滴后加水溶清;
13.s3、再加入甲基叔丁基醚搅拌分成,水层用甲基叔丁基醚萃取一次,合并有机相过滤,滤液减压浓缩;
14.s4、浓缩液开始水解,向浓缩液中加入15%碱性水溶液,甲醇,常温搅拌互溶;
15.s5、水解完毕后开始减压浓缩甲醇。浓缩干后体系开始分层,上层有机相,下层水
相;
16.s6、水相再用二氯甲烷萃取两次,合并有机相减压浓缩得到5-己烯-1-醇。
[0017][0018]
在本发明中可选的,所述四丁基溴化铵可以用四丁基氯化铵替代。
[0019]
在本发明中可选的,所述甲基叔丁基醚可以用二氯甲烷、乙酸乙酯替代。
[0020]
在本发明中可选的,所述碱性水溶液包括氢氧化钠水溶液,氢氧化钾水溶液,氨水溶液。
[0021]
在本发明中可选的,所述6-溴-1-己烯:四丁基溴化铵:醋酸钾:甲基叔丁基醚:碱性水溶液的比例为10~15:1:12~20:22~29:10~15。
[0022]
在本发明中可选的,所述s1中混合的反应液加热温度为78-83℃,反应时间为2小时。
[0023]
在本发明中可选的,所述s1中制得产品为5-烯己酯。
[0024]
本发明提供了合成5-己烯-1-醇的方法,具备以下有益效果:
[0025]
1、该合成5-己烯-1-醇的方法,通过以6-溴-1-己烯为原料,四丁基溴化铵作为催化剂,所使用的方法反应条件简单,反应时间短,工艺简单,容易实现,成本较低,适合工业化生产5-己烯-1-醇的合成方法,并且其中乙腈是有机实验室最常用的有机溶剂之一,醋酸钾是一种价格低廉的原料,反应条件简单,制得的5-己烯-1-醇纯度高,反应后处理非常简单,大大降低了生产成本,具有更强的经济实用性和灵活性。
[0026]
2、该合成5-己烯-1-醇的方法,通过使用6-溴-1-己烯为原料,能够有效避开方法一中5-己烯酸甲酯或者5-己烯酸用强金属还原剂进行还原和方法四中5-己炔-1-醇通过加氢反应存在的问题,避免过程中产生氢气和使用氢气,加氢本身属于高危工艺操作,安全风险高,容易产生生产意外;也可以避免方法二中使用1,6-己二醇通过300℃高温消除,反应副产物多,对设备要求苛刻的问题,相比于目前使用的5-己烯-1-醇的合成方法而言,本发明合成5-己烯-1-醇的方法条件非常温和,能够极大的提高安全性和可操作性,方便迅速进行工业化生产。
附图说明
[0027]
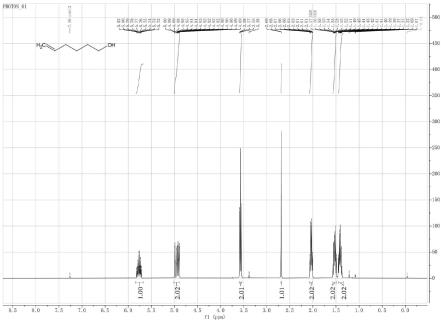
图1为本发明的核磁氢谱
[0028]
图2为本发明实施例1的示意图;
[0029]
图3为本发明实施例2的示意图;
[0030]
图4为本发明实施例3的示意图;
[0031]
图5为本发明实施例4的示意图。
具体实施方式
[0032]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。
[0033]
请参阅图1-图5,本发明提供技术方案:合成5-己烯-1-醇的方法,
[0034]
实施例1
[0035]
将200.0g6-溴-1-己烯,39.4g四丁基溴化铵溶于400ml乙腈中,加入144.0g醋酸钾,混合液加热到82℃回流,2小时后检测反应完全。将混合液冷却至20℃,然后减压浓缩干,向浓缩物中加入400ml水溶清,再加入200ml甲基叔丁基醚搅拌分层,水相用100ml甲基叔丁基醚萃取一次。合并有机相,干燥,体系过滤后滤液减压浓缩干后得到5-烯己酯。
[0036]
在25℃下,将5-烯己酯溶于300ml的甲醇溶液中,加入331.5g的15%氢氧化钠,反应2小时检测反应完全。体系开始浓缩甲醇,浓缩干后体系开始分层,上层有机相,下层水相。水相再用二氯甲烷萃取两次,合并有机相,减压浓缩,浓缩干后得到目标产物。产率达到82.4%,纯度99.7%。(10:1:12:22:10)
[0037]
实施例2
[0038]
将60.0g6-溴-1-己烯,7.88g四丁基溴化铵溶于80ml乙腈中,加入48.02g醋酸钾,混合液加热到82℃回流,2小时后检测反应完全。将混合液冷却至20℃,然后减压浓缩干,向浓缩物中加入80ml水溶清,再加入40ml甲基叔丁基醚搅拌分层,水相用40ml甲基叔丁基醚萃取一次。合并有机相,干燥,体系过滤后滤液减压浓缩干后得到5-烯己酯。
[0039]
在25℃下,将5-烯己酯溶于70ml的甲醇溶液中,加入98.0g的15%氢氧化钠,反应2小时检测反应完全。体系开始浓缩甲醇,浓缩干后体系开始分层,上层有机相,下层水相。水相再用二氯甲烷萃取两次,合并有机相,减压浓缩,浓缩干后得到目标产物。产率达到82.0%,纯度99.3%。(15:1:20:30:15)
[0040]
实施例3
[0041]
将200.0g6-溴-1-己烯,34.19g四丁基氯化铵溶于400ml乙腈中,加入144.0g醋酸钾,混合液加热到82℃回流,2小时后检测反应完全。将混合液冷却至20℃,然后减压浓缩干,向浓缩物中加入400ml水溶清,再加入200ml甲基叔丁基醚搅拌分层,水相用100ml甲基叔丁基醚萃取一次。合并有机相,干燥,体系过滤后滤液减压浓缩干后得到5-烯己酯。
[0042]
在25℃下,将5-烯己酯溶于300ml的甲醇溶液中,加入462.93g的15%氢氧化钾,反应2小时检测反应完全。体系开始浓缩甲醇,浓缩干后体系开始分层,上层有机相,下层水相。水相再用二氯甲烷萃取两次,合并有机相,减压浓缩,浓缩干后得到目标产物。产率达到78%,纯度99.8%。(10:1:12:22:10)
[0043]
实施例4
[0044]
将200.0g6-溴-1-己烯,39.4g四丁基溴化铵溶于400ml乙腈中,加入144.0g醋酸钾,混合液加热到82℃回流,2小时后检测反应完全。将混合液冷却至20℃,然后减压浓缩干,向浓缩物中加入400ml水溶清,再加入200ml二氯甲烷搅拌分层,水相用90ml二氯甲烷萃取一次。合并有机相,干燥,体系过滤后滤液减压浓缩干后得到5-烯己酯。
[0045]
在25℃下,将5-烯己酯溶于300ml的甲醇溶液中,加入331.5g的15%氢氧化钠,反应2小时检测反应完全。体系开始浓缩甲醇,浓缩干后体系开始分层,上层有机相,下层水相。水相再用二氯甲烷萃取两次,合并有机相,减压浓缩,浓缩干后得到目标产物。产率达到76%,纯度99.1%。(10:1:12:22:10)
[0046]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1