一种双哌啶的合成方法与流程
1.本发明涉及化工领域,具体涉及一种双哌啶的合成方法。
背景技术:
2.手性双哌啶的合成在催化领域中意义重大,早在1851年,stenhouse通过分离首次获得了金雀花碱(-)-sparteine。(-)-sparteine是自然界存在的一种羽扇豆生物碱,作为手性诱导双氮配体广泛应用于不对称催化反应领域。如(-)-sparteine可用于pd催化仲醇需氧氧化动力学拆分反应、吡咯烷不对称锂化/去质子反应、pd-催化的保持构型的烯丙基化反应、亚胺与有机锂加成反应等不同催化反应中。(-)-sparteine是天然生物碱,资源极其有限,人工全合成又十分困难,而且在研究(-)-sparteine的过程中又发现,在自然界中(-)-sparteine是单极存在的,并不存在对映体,不仅通过拆分不能得到,而且通过衍生对它的结构进行修饰也很难。研究发现双哌啶环是(-)-sparteine重要的中心结构,在桥环上没有取代基,并且它的结构刚性很强。由于(-)-sparteine的良好的催化性能,科学家们希望能通过合成类似结构的化合物代替或优化(-)-sparteine构成的手性催化体系。
3.(-)-sparteine是一种天然生物碱,具有和好的手性诱导作用,因此广泛应用于不对称催化反应领域。stenhouse在1851年首次成功分离出(-)-spartein。自此,很多人致力于对金雀花碱的研究。(-)-sparteine具有很强的刚性结构,其核心部分是笼状的双哌啶结构。但金雀花碱作为天然产物,资源有限,全合成难度较大。因此,如何用简单、环保的化工方法合成双哌啶是一个重要的技术问题。
技术实现要素:
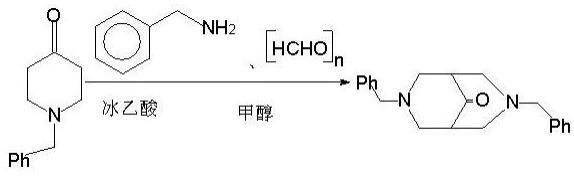
4.针对上述技术问题,本发明提供一种双哌啶的合成方法,采取的技术方案为:一种双哌啶的合成方法,其包括以下步骤:(1)双苄基双哌啶酮的合成:将冰乙酸和甲醇混合后,冰水浴冷却下滴加到有苄基哌啶酮和甲醇的三口瓶中,得混合物a;氮气保护下,在四口瓶中加入甲醇和苄胺,用冰水浴控制温度,滴加冰乙酸,滴加完毕后加入多聚甲醛,用水浴控制温度,向该混合物滴加上述混合物a,滴加完毕,反应进程用gc检测跟踪,待反应组分不再有明显变化后,停止加热,冷却过滤,滤液旋蒸浓缩,残留物加冷水用乙醚萃取,然后用二氯甲烷萃取,萃取液用无水硫酸钠干燥,过滤,旋蒸浓缩得酒红色粘稠油状液体,用乙酸乙酯和正己烷重结晶,过滤得白色晶体,即为双苄基双哌啶酮;(2)双苄基双哌啶的合成:氮气保护下,向四口瓶中依次加入koh、一缩二乙二醇、步骤(1)所得双苄基双哌啶酮及水合肼,加热回流后,蒸出肼和水的混合物至第一反应温度再回流,继续蒸出肼和水的混合物至第二反应温度再回流,停止反应并冷却至室温后,加冷水,用乙醚萃取,萃取液依次用氢氧化钠溶液及饱和食盐水溶液洗涤,然后用无水硫酸钠干燥,过滤除溶剂,滤液蒸出溶剂得黄色粘稠液体,减压蒸馏得无色透明液体,即为双苄基双哌啶;
(3)双哌啶的合成:在氮气保护下,向装有磁力搅拌的三口瓶中加入pd/c催化剂、乙酸及步骤(2)所得双苄基双哌啶,控制水浴温度,搅拌下通氢气,tlc检测至反应完全后过滤,滤液旋蒸浓缩,浓缩液在冰水冷却下用乙醚萃取,萃取液用无水硫酸钠干燥,过滤除去干燥剂,滤液旋蒸除溶剂得淡黄色油状物,减压蒸馏得无色透明液体,即为双哌啶。
5.2.根据权利要求1所述的一种双哌啶的合成方法,其特征在于,所述步骤(1)苄基哌啶酮为16-19ml,苄胺8-12为ml,多聚甲醛为10-14g;冰乙酸先后向苄基哌啶酮滴加时为5.0-6.5ml、向苄胺滴加时为4-7ml;甲醇先后与冰乙酸混合时为50-70ml、与苄基哌啶酮混合时为100-200ml、与苄胺混合时为100-200ml。
6.所述步骤(1)用冰水浴控制温度为5-10℃;用水浴控制温度为40-45℃;残留物加冷水用乙醚萃取时水相用质量分数为31%的koh调节调ph值为12-14。
7.所述步骤(1)所得白色晶体即双苄基双哌啶酮为15-17g,产率为45-55%,mp.60-80℃。
8.所述步骤(2)koh的质量分数为80%,加入量为10-15g;一缩二乙二醇加入量为50-80ml;双苄基双哌啶加入量为5-8g;水合肼的质量分数为80%,加入量为4-8g;冷水加入量为40-60ml。
9.所述步骤(2)加热回流3-5小时;第一反应温度为140-160℃,再回流2-4小时;第二反应温度为180-200℃,再回流3-5小时。
10.所述步骤(2)所得无色透明液体即双苄基双哌啶为4.0-5.0g,产率为90-95%。
11.所述步骤(3)pd/c催化剂的质量分数为10%,加入量为0.5-3g;乙酸的质量分数为85%,加入量为10-30ml;双苄基双哌啶加入量为3-6g。
12.所述步骤(3)控制水浴温度为20-30℃;浓缩液冰水冷却时,用质量分数为40%的koh溶液调节调ph值为11-12。
13.所述步骤(3)所得无色透明液体即双哌啶2-3g,gc纯度为89-93%,产率为45-55%。
14.本发明主要具有以下有益技术效果:1.本发明通过双苄基双哌啶酮的合成、双苄基双哌啶的合成、双哌啶的合成这三个步骤构成一条流畅的合成路线,方法简单,容易推广。
15.2.本发明原料易得,生产过程不产生有害废物,具有良好的环保性。
16.3.本发明采用气相色谱法(gc)检测反应进程,判断反应终点,并且对合成过程中的中间化合物及目标产物的结构通过红外、核磁表征进行了定性分析,确保合成反应的精准性。
17.4.本发明合成率高。据测定,双哌啶的gc纯度达89-93%,产率达45-55%。
附图说明
18.图1为本发明的双苄基双哌啶酮的合成工艺简图;图2为本发明的双苄基双哌啶的合成工艺简图;图3为本发明的双哌啶的合成工艺简图。
具体实施方式
19.下面结合具体实施例及附图对本发明作进一步描述,在此发明的示意性实施例以
及说明用来解释本发明,但并不作为对本发明的限定。
20.实施例1一种合双哌啶的合成方法,包括以下步骤:如图1,双苄基双哌啶酮的合成:将5.7ml(0.1mol)冰乙酸和60ml甲醇混合后,冰水浴冷却下滴加到有苄基哌啶酮17.9ml(0.1mol)和150ml甲醇的500ml三口瓶中,得混合物a;氮气保护下,在四口瓶中加入甲醇150ml,苄胺10.9ml(0.1mol),用冰水浴控制温度8℃,在此温度下,滴加冰乙酸5.7ml(0.1mol),滴加完毕,加入多聚甲醛12.6g(0.42mol),水浴控制温度42℃,向该混合物滴加上述混合物a,滴加完毕,反应进程用gc检测跟踪,待反应组分不再有明显变化后,停止加热,冷却过滤,滤液旋蒸浓缩,残留物加冷水用乙醚萃取,水相用质量分数为31%的koh调节ph值为13,然后用二氯甲烷萃取,萃取液用无水硫酸钠干燥,过滤,旋蒸浓缩得酒红色粘稠油状液体,用乙酸乙酯和正己烷重结晶,过滤得白色晶体即双苄基双哌啶酮16.34g,产率为51%,mp.79℃。
21.如图2,双苄基双哌啶的合成:氮气保护下,向四口瓶中依次加入12.00g(0.176mol)质量分数为82%的koh、一缩二乙二醇70ml、6.00g(0.022mol)的双苄基双哌啶酮以及5.5g(0.088mol)质量分数为80%的水合肼,加热回流4小时后,蒸出肼和水的混合物至反应温度至150℃,再回流3小时,继续蒸出肼和水的混合物至反应温度至190℃,再回流4小时,停止反应并冷却至室温后,加冷水50ml,用乙醚萃取(30ml*5),萃取液依次用氢氧化钠溶液(30ml*3)、饱和食盐水溶液(30ml*3)洗涤,然后用无水硫酸钠干燥,过滤除溶剂,滤液蒸出溶剂得黄色粘稠液体,减压蒸馏得无色透明液体即双苄基双哌啶4.65g,产率94%。
22.如图3,双哌啶的合成:在氮气保护下,向装有磁力搅拌的100ml三口瓶中加入0.5g质量分数为10%的pd/c催化剂、15ml质量分数为85%的乙酸及3.80(0.0147mmol)的双苄基双哌啶,控制水浴温度25℃,搅拌下通氢气,tlc检测至反应完全后过滤,滤液旋蒸浓缩,浓缩液在冰水冷却下用质量分数为40%的koh溶液调节ph值为11.5,乙醚萃取,萃取液用无水硫酸钠干燥,过滤除去干燥剂,滤液旋蒸除溶剂得淡黄色油状物,减压蒸馏得无色透明液体即双哌啶2.0g,gc纯度90%,产率50%。
23.实施例2一种合双哌啶的合成方法,包括以下步骤:如图1,双苄基双哌啶酮的合成:将5.0ml冰乙酸和50ml甲醇混合后,冰水浴冷却下滴加到有苄基哌啶酮16ml和100ml甲醇的500ml三口瓶中,得混合物a;氮气保护下,在四口瓶中加入甲醇100ml,苄胺8ml,用冰水浴控制温度5℃,在此温度下,滴加冰乙酸4ml,滴加完毕,加入多聚甲醛10g,水浴控制温度40℃,向该混合物滴加上述混合物a,滴加完毕,反应进程用gc检测跟踪,待反应组分不再有明显变化后,停止加热,冷却过滤,滤液旋蒸浓缩,残留物加冷水用乙醚萃取,水相用质量分数为31%的koh调节ph值为12,然后用二氯甲烷萃取,萃取液用无水硫酸钠干燥,过滤,旋蒸浓缩得酒红色粘稠油状液体,用乙酸乙酯和正己烷重结晶,过滤得白色晶体即双苄基双哌啶酮15.2g,产率45%,mp.60℃。
24.如图2,双苄基双哌啶的合成:氮气保护下,向四口瓶中依次加入10g质量分数为82%的koh、一缩二乙二醇50ml、5g的双苄基双哌啶酮以及4g质量分数为80%的水合肼,加热回流3小时后,蒸出肼和水的混合物至反应温度至140℃,再回流3小时,继续蒸出肼和水的混合物至反应温度至180℃,再回流3小时,停止反应并冷却至室温后,加冷水40ml,用乙醚
萃取(30ml*5),萃取液依次用氢氧化钠溶液(30ml*3)、饱和食盐水溶液(30ml*3)洗涤,然后用无水硫酸钠干燥,过滤除溶剂,滤液蒸出溶剂得黄色粘稠液体,减压蒸馏得无色透明液体即双苄基双哌啶4.05g,产率90%。
25.如图3,双哌啶的合成:在氮气保护下,向装有磁力搅拌的100ml三口瓶中加入1.5g质量分数为10%的pd/c催化剂、10ml质量分数为85%的乙酸及3g的双苄基双哌啶,控制水浴温度20℃,搅拌下通氢气,tlc检测至反应完全后过滤,滤液旋蒸浓缩,浓缩液在冰水冷却下用质量分数为40%的koh溶液调节ph值为11,乙醚萃取,萃取液用无水硫酸钠干燥,过滤除去干燥剂,滤液旋蒸除溶剂得淡黄色油状物,减压蒸馏得无色透明液体即双哌啶2.5g,gc纯度89%,产率45%。
26.实施例3一种合双哌啶的合成方法,包括以下步骤:如图1,双苄基双哌啶酮的合成:将6.5ml冰乙酸和70ml甲醇混合后,冰水浴冷却下滴加到有苄基哌啶酮19ml和200ml甲醇的500ml三口瓶中,得混合物a;氮气保护下,在四口瓶中加入甲醇200ml,苄胺12ml,用冰水浴控制温度10℃,在此温度下,滴加冰乙酸7ml,滴加完毕,加入多聚甲醛14g,水浴控制温度45℃,向该混合物滴加上述混合物a,滴加完毕,反应进程用gc检测跟踪,待反应组分不再有明显变化后,停止加热,冷却过滤,滤液旋蒸浓缩,残留物加冷水用乙醚萃取,水相用质量分数为31%的koh调节ph值为14,然后用二氯甲烷萃取,萃取液用无水硫酸钠干燥,过滤,旋蒸浓缩得酒红色粘稠油状液体,用乙酸乙酯和正己烷重结晶,过滤得白色晶体即双苄基双哌啶酮16.95g,产率55%,mp.80℃。
27.如图2,双苄基双哌啶的合成:氮气保护下,向四口瓶中依次加入15g质量分数为82%的koh、一缩二乙二醇80ml、8g的双苄基双哌啶酮以及8g质量分数为80%的水合肼,加热回流5小时后,蒸出肼和水的混合物至反应温度至160℃,再回流3小时,继续蒸出肼和水的混合物至反应温度至200℃,再回流5小时,停止反应并冷却至室温后,加冷水60ml,用乙醚萃取(30ml*5),萃取液依次用氢氧化钠溶液(30ml*3)、饱和食盐水溶液(30ml*3)洗涤,然后用无水硫酸钠干燥,过滤除溶剂,滤液蒸出溶剂得黄色粘稠液体,减压蒸馏得无色透明液体即双苄基双哌啶5.02g,产率95%。
28.如图3,双哌啶的合成:在氮气保护下,向装有磁力搅拌的100ml三口瓶中加入3g质量分数为10%的pd/c催化剂、30ml质量分数为85%的乙酸及6g的双苄基双哌啶,控制水浴温度30℃,搅拌下通氢气,tlc检测至反应完全后过滤,滤液旋蒸浓缩,浓缩液在冰水冷却下用质量分数为40%的koh溶液调节ph值为12,乙醚萃取,萃取液用无水硫酸钠干燥,过滤除去干燥剂,滤液旋蒸除溶剂得淡黄色油状物,减压蒸馏得无色透明液体即双哌啶2.98g,gc纯度93%,产率55%。
29.本发明采用气相色谱法(gc)检测反应进程,判断反应终点,并且对合成过程中的中间化合物及目标产物的结构通过红外、核磁表征进行了定性分析。上述实施例合成工艺参见图1、图2及图3。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1