噻唑橙有机硅季铵盐化合物及其制备方法和应用与流程
1.本发明涉及用于纺织品的抗菌染料技术领域,涉及一种噻唑橙有机硅季铵盐化合物及其制备方法和应用。
背景技术:
2.抗菌染料是一类同时具有染色功能和抗菌功能的多功能化合物,在医疗、服饰、食品等行业都有着巨大的应用前景。例如,织物的染色和抗菌整理在传统上通常都是两个独立的环节。而这样的处理方法会存如下问题:一方面,某些抗菌性整理有可能会影响染色性能,或者其他的功能性整理; 另一方面,抗菌性整理往往需要单独的后处理过程,分步进行染色和整理会导致成本的增加和资源的浪费,也会造成更大的环境破坏,不符合可持续发展的要求。而使用抗菌染料则可以将织物的染色和抗菌整理在同一个步骤里同时进行,在对织物进行染色的同时就赋予织物以抗菌性能。相较传统工艺,使用抗菌染料减少了一步湿处理过程,很好地降低了能耗,并减少了用水量和废水排放量,无疑更加绿色环保。
3.噻唑橙是一类分子结构中同时含有噻唑环和喹啉环的阳离子染料,其具有颜色鲜明,荧光量子产率高,摩尔消光系数大等诸多特点。噻唑橙常被用作核酸的荧光探针,在电泳分离、dna的定量测定等领域得到广泛应用。噻唑橙的阳离子结构特性,使其具有与其他阳离子型抗菌剂进行组合使用的潜质。以噻唑橙为母核来设计一系列的抗菌染料化合物,将是一项非常有意义的工作。
技术实现要素:
4.鉴于上述现有技术中存在的问题,本发明开发了一种噻唑橙有机硅季铵盐化合物及其制备方法,以及这种噻唑橙有机硅季铵盐化合物在抗菌领域的应用。
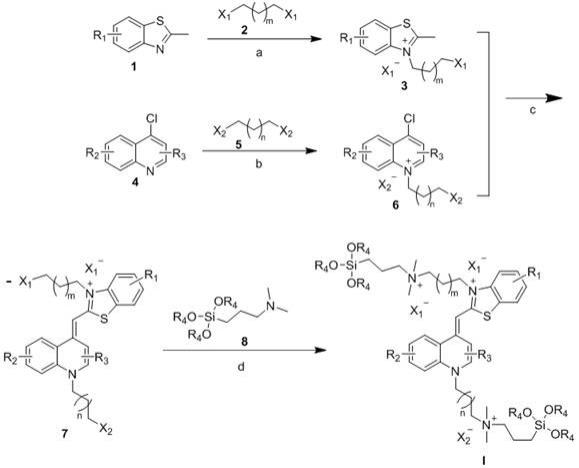
5.本发明的技术方案之一是提供一种噻唑橙有机硅季铵盐化合物,上述化合物结构如式i所示,其中,x1和x2选自氯、溴、碘、对甲苯磺酰氧基、苯磺酰氧基、甲磺酰氧基, r1,r2,r3,r4分别独立选自甲基、乙基、c3-c8烷基、苯基、硝基、亚硝基、磺酰基、甲磺酰基、氟、氯、溴、碘、三氟甲基、二氟甲基, m=0-30,n=0-30。
6.在本发明的一较佳实施例中,上述的m大于等于8,并小于等于12;上述的n大于等于10,并小于等于15。
7.本发明的技术方案之二是提供一种上述噻唑橙有机硅季铵盐化合物的制备方法,包括如下步骤:(a) 在隔绝空气的条件下,反应釜中加入第一反应溶剂,式1化合物,式2化合物,反应温度60-200℃,反应时间为6-72小时,得式3化合物,其中,上述式样1化合物和式2化合物的物质的量比为1:1-10; (b)隔绝空气的条件下,反应釜中加入第二反应溶剂,然后加入式4化合物和式5化合物,反应温度60-200℃,反应时间3-72小时,得式6化合物,其中,上述式样3化合物和式4化合物的物质的量比为1:1-10;(c)在隔绝空气的条件下,反应釜中加入第三反应溶剂,式3化合物和式\6化合物,反应温度25-60℃反应,保温反应1-5小时,得式7化合物,其中,上述式3化合物和式6化合物的物质的量比为1:1-10; (d)在隔绝空气的条件下,反应釜中加入第四反应溶剂,式7化合物和式8化合物,反应温度80-200℃反应,保温反应12-102小时,得式i化合物,其中,上述式7化合物和式8化合物的物质的量比为1:1-10;其中,x1和x2选自氯、溴、碘、对甲苯磺酰氧基、苯磺酰氧基、甲磺酰氧基, r1,r2,r3,r4分别独立选自甲基、乙基、c3-c8烷基、苯基、硝基、亚硝基、磺酰基、甲磺酰基、氟、氯、溴、碘、三氟甲基、二氟甲基, m=0-30,n=0-30。
8.在本发明的一较佳实施例中,上述的m大于等于8,并小于等于12;上述的n大于等于10,并小于等于15。
9.在本发明的一较佳实施例中,步骤(a)中,上述第一反应溶剂为乙腈,对二甲苯,邻
二甲苯,间二甲苯,甲苯,乙二醇二乙醚,乙二醇二甲醚,dmf,dmso,乙腈,乙酸乙酯,四氢呋喃,2-甲基四氢呋喃或者二氧六环中的至少一种。
10.在本发明的一较佳实施例中,步骤(b)中,上述第二反应溶剂为乙腈,对二甲苯,邻二甲苯,间二甲苯,甲苯,乙二醇二乙醚,乙二醇二甲醚,dmf,dmso,乙酸乙酯,四氢呋喃,2-甲基四氢呋喃或者二氧六环中的至少一种。
11.在本发明的一较佳实施例中,步骤(c)中,上述第三反应溶剂为乙醇,甲醇,异丙醇,正丁醇、对二甲苯,邻二甲苯,间二甲苯,甲苯,乙二醇二乙醚,乙二醇二甲醚,dmf,dmso,乙腈,四氢呋喃,2-甲基四氢呋喃,二氧六环中的至少一种。
12.在本发明的一较佳实施例中,上述第四反应溶剂为邻二甲苯,对二甲苯,间二甲苯,甲苯,乙二醇二乙醚, dmf,dmso,乙腈或者丙酮中的至少一种。
13.在本发明的一较佳实施例中,步骤(d)中,还包括需要继续加入碱性试剂,所述的碱性试剂为三乙胺,n,n-二异丙基乙胺中的至少一种。
14.本发明的技术方案之三是提供上述的噻唑橙有机硅季铵盐化合物和采用上述噻唑橙有机硅季铵盐化合物的制备方法制得噻唑橙有机硅季铵盐化合物在抗菌材料中的应用。
15.有益效果本发明的噻唑橙有机硅季铵盐化合物具有噻唑环,喹啉环,季铵基和有机硅氧烷四种结构,抗菌谱广。本发明对金黄色葡萄球菌,大肠杆菌,白色念球菌和黑曲霉菌均具有抗菌活性。
16.本发明的耐洗性尤其好,100次洗涤后的抑菌率都保持在99.9%,200次洗涤后的抑菌率都在80%以上,300次洗涤后的抑菌率也都还在70%以上。
17.与现有制备方法相比,本发明操作简单,原料来源方便,对环境友好,适合工业化生产,具有良好的应用前景。
具体实施方式
18.下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
19.实施例1:
(m,2h)。
27.步骤(c):式7-1化合物的制备保持氮气微正压,向5 l反应瓶中依次加入1.2 l 乙醇,258.41g(0.736mol)式3-1化合物和 269.00 g(0.436 mol)式6-1化合物,搅匀。然后向其中加入7.45 g(0.074mol)三乙胺。加完后,搅匀,反应液升温至40 ℃反应。保温反应3小时,反应完成。
28.反应液冷却至室温,向其中加入1l乙酸乙酯和1l石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到391.19g(0.653 mol)红色固体产品。
29.收率88.70%。
30.产品纯度:99.25%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.50(d,1h),8.37(d,1h),8.25-8.30(m,3h),7.74-7.93(m,5h),6.87(s,1h),4.77-4.89 (m,2h),3.73(t,2h),3.21(t,2h),3.05-3.21 (m,2h),2.39-2.49(m,2h),2.01-2.05(m,2h)。
31.步骤(d):式i-1化合物的制备保持氮气微正压,向5 l反应瓶中依次加入1.8l 邻二甲苯,g(0.632 mol)式7-1化合物和g(1.355 mol)(2-10 eq)(n,n-二甲基-3-氨丙基)三甲氧基硅烷,加完后,搅匀;反应液升温至140 ℃反应。保温反应8小时,反应完成。
32.反应液自然冷却至室温,然后继续冷却至0℃,保温析晶3小时,析出大量固体,过滤,收集滤饼得到594.39g(0.586mol)红色固体产品。
33.收率89.8%,4步反应总收率40.74%,产品纯度:99.21%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.53(d,1h),8.36(d,1h),8.25-8.30(m,3h),7.74-7.93(m,5h), 6.93(s,1h),4.79-4.92(m,2h),3.52(bs,18h),3.05-3.25 (m,22h),2.23-2.32(m,2h),1.96-2.01(m,2h),1.72-1.77 (m,4h),0.65(t,4h)。
34.实施例2:
步骤(a):式3-2化合物的制备保持氮气微正压,向5 l反应瓶中依次加入2l 2-甲基四氢呋喃, 149.21 g(1.0 mol)式1-2化合物2-甲基苯并噻唑, 282.08g(2.0 mol)式2-2化合物1,5-二氯戊烷。加完后,搅匀,反应液升温至80 ℃反应。保温反应25小时,反应完成。
35.反应液冷却至室温,向其中加入3000 ml石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到208.4g(0.718 mol)黄色固体产品。
36.收率71.8%。
37.产品纯度:99.13%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.51(d,1h),8.37(d,1h),7.91(t,1h),7.82(t,1h),4.75-4.85 (m,2h),3.58(t,2h),3.25(s,3h),2.01-2.03(m,2h),1.75-1.79(m,2h),1.27-1.30(m,2h)。
38.步骤(b):式6-2化合物的制备保持氮气微正压,向20 l反应瓶中依次加入6l乙二醇二乙醚,532.89 g(3.0 mol)式4-2化合物2-甲基-4-氯喹啉,1856.52g(6.0mol)式5-2化合物1,4-二碘丁烷。加完后,搅匀,反应液升温至120 ℃反应。保温反应10小时,反应完成。
39.反应液冷却至室温,向其中加入3000 ml石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到1004.84(2.061mol)黄色固体产品。
40.收率68.7%。
41.产品纯度:98.95%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.56(d,1h),8.46(d,1h),8.22(t,1h),8.01(t,1h),7.55(s,1h),5.18-5.25(m,2h),2.93(t,2h),2.67(s,3h),2.00-2.03(m,2h),1.82-1.85(m,2h)步骤(c):式7-2化合物的制备保持氮气微正压,向5 l反应瓶中依次加入1.2 l 乙醇,208.4g(0.718 mol)式3-2化合物和700.12g(1.436mol)式6-2化合物,搅匀。然后向其中加入14.53g(0.144mol)三乙胺。加完后,搅匀,反应液升温至40 ℃反应。保温反应4小时,反应完成。
42.反应液冷却至室温,向其中加入1l乙酸乙酯和1l石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到395.04g(0.644 mol)红色固体产品。
43.收率89.7%。
44.产品纯度:99.46%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.50(d,1h),8.37(d,1h),8.25-8.30(m,3h),7.74-7.93(m,5h),6.87(s,1h),4.77-4.89 (m,2h),3.73(t,2h),3.21(t,2h),3.05-3.21 (m,2h),2.39-2.49(m,2h),2.01-2.05(m,2h)。
45.步骤(d):式i-2化合物的制备保持氮气微正压,向5 l反应瓶中依次加入1.8l 邻二甲苯, 395.04g(0.644 mol)式7-2化合物和481.93g(1.932 mol)(3 eq)3
‑ꢀ
n,n-二甲基氨丙基(三乙氧基)硅烷(cas:43108-00-5),加完后,搅匀;反应液升温至140 ℃反应。保温反应8小时,反应完成。
46.反应液自然冷却至室温,然后继续冷却至0℃,保温析晶3小时,析出大量固体,过滤,收集滤饼得到650.45g(0.585mol)红色固体产品。
47.收率90.8%,4步反应总收率40.18%,产品纯度:98.64%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.53(d,1h),8.36(d,1h),8.25-8.30(m,3h),7.74-7.93(m,5h),6.93(s,1h),4.79-4.92(m,2h),3.85(q,12h),3.05-3.25 (m,22h),2.23-2.32(m,2h),1.96-2.01(m,2h),1.73-1.78(m,4h),1.22(t,18h),0.64(t,4h)。
48.实施例3:
步骤(a):式3-3化合物的制备保持氮气微正压,向5 l反应瓶中依次加入2l乙二醇二乙醚,149.21 g(1.0 mol)式1-1化合物2-甲基-6-硝基苯并噻唑, 1279.62 g(3.0 mol)(1-10 eq)式2-3化合物1,6-二对甲苯磺酰氧基己烷。加完后,搅匀,反应液升温至120℃(60-200)反应。保温反应18小时,反应完成。
49.反应液冷却至室温,向其中加入2000 ml石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到430.80g(0.694 mol)绿色固体产品。
50.收率70.9%。产品纯度:98.87%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 9.45(s,1h),8.67(d,1h),8.52(d,1h),7.79(d,2h),7.47(d,2h),7.34(d,2h),7.10(d,2h),4.80-4.93(m,2h),4.02(t,2h),3.28(s,3h),2.45(s,3h),2.28(s,3h),2.02-2.04(m,2h),1.62-1.66(m,2h),1.42-1.45(m,2h),1.22-1.27(m,2h)。
51.步骤(b):式6-3化合物的制备保持氮气微正压,向20l反应瓶中依次加入6l dmf,507.16 g(3.1mol)式4-3化合物4-氯喹啉,1442.06g(9.3mol)式5-3化合物1,6-二氯己烷。加完后,搅匀,反应液升温至150℃反应。保温反应8小时,反应完成。
52.反应液冷却至室温,向其中加入6000 ml石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到681.73g(2.140mol)黄色固体产品。
53.收率69.01%。产品纯度:98.89%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.49-8.74(m,2h),8.31-8.41(m,2h),δ 8.06-8.21(m,2h), 5.29-5.36(m,2h),3.56(t,2h),2.49
–
2.57 (m,2h),2.01-2.03(m,2h),1.75-1.79(m,2h),1.45-1.48(m,2h),1.27-1.30(m,2h)。
54.步骤(c):式7-3化合物的制备保持氮气微正压,向5 l反应瓶中依次加入1.2 l 乙醇,430.80g(0.694 mol)式3-3化合物和 663.47 g(2.082mol)式6-3化合物,搅匀。然后向其中加入21.07g(0.208 mol)三乙胺。加完后,搅匀,反应液升温至40 ℃反应。保温反应5小时,反应完成。
55.反应液冷却至室温,向其中加入1l乙酸乙酯和1l石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到542.42g(0.626mol)橙色固体产品。
56.收率90.2%。
57.产品纯度:98.36%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 9.45(s,1h),8.67(d,1h),8.52(d,1h),8.25-8.30(m,3h),7.79(d,2h),7.65-7.70(m,2h),7.47(d,2h),7.34(d,2h),7.31(d,1h),7.15(s,1h),7.10(d,2h),4.80-4.93(m,2h),4.02(t,2h),3.57(d,2h),3.52(bs,18h),3.25-3.38(m,2h),2.45(s,3h),2.28(s,3h),2.01-2.04(m,4h),1.75-1.79(m,2h),1.62-1.66(m,2h),1.42-1.45(m,2h),1.22-1.27(m,6h)。
58.步骤(d):式i-3化合物的制备保持氮气微正压,向5 l反应瓶中依次加入1.8l 邻二甲苯, 542.42g(0.626mol)式7-3化合物和389.40g(1.878mol)(2-10 eq)(n,n-二甲基-3-氨丙基)三甲氧基硅烷,加完后,搅匀;反应液升温至140 ℃反应。保温反应10小时,反应完成。
59.反应液自然冷却至室温,然后继续冷却至0℃,保温析晶3小时,析出大量固体,过滤,收集滤饼得到729.03 g(0.569mol)橙色固体产品。
60.收率90.9%,4步反应总收率39.27%,产品纯度:98.74%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 9.49(s,1h),8.71(d,1h),8.54(d,1h),8.25-8.30(m,3h),7.65-7.70(m,2h), 7.47(d,4h),7.32(d,1h),7.17(s,1h),7.10(d,4h),4.80-4.93(m,2h),3.25-3.38(m,2h),3.12(bs,20h),2.28(s,6h),2.01-2.04(m,4h),1.72-1.77 (m,4h),1.55-1.59(m,4h),1.22-1.30(m,8h),0.64(t,4h)。
61.实施例4:
5.38(m,2h),3.84(t,2h),2.50
–
2.58 (m,2h)。
69.步骤(c):式7-4化合物的制备保持氮气微正压,向5 l反应瓶中依次加入2.0 l 乙醇,190.87(0.728 mol)式3-4化合物和 805.40 g(2.912mol)式6-4化合物,搅匀。然后向其中加入29.47 g(0.291mol)三乙胺。加完后,搅匀,反应液升温至40 ℃反应。保温反应3小时,反应完成。
70.反应液冷却至室温,向其中加入1l乙酸乙酯和1l石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到301.50g(0.647 mol)红色固体产品。
71.收率88.9%。
72.产品纯度:99.31%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.48(d,1h),8.36(d,1h),8.25-8.31(m,3h),7.74-7.92(m,5h),6.87(s,1h),4.77-4.89 (m,2h),3.73(t,2h),3.21(t,2h),3.05-3.21 (m,2h),2.39-2.49(m,2h),2.01-2.04(m,2h)。
73.步骤(d):式i-4化合物的制备保持氮气微正压,向5 l反应瓶中依次加入1.8l 邻二甲苯,301.50g(0.647 mol)式7-4化合物和645.72g(2.589 mol)3
‑ꢀ
n,n-二甲基氨三乙氧基硅烷(cas:43108-00-5),加完后,搅匀;反应液升温至140 ℃反应。保温反应8小时,反应完成。
74.反应液自然冷却至室温,然后继续冷却至0℃,保温析晶3小时,析出大量固体,过滤,收集滤饼得到553.18 g(0.573mol)红色固体产品。
75.收率88.60%,4步反应总收率39.11%,产品纯度:98.89%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.52(d,1h),8.37(d,1h),8.24-8.29(m,3h),7.74-7.93(m,5h),6.93(s,1h),4.79-4.93(m,2h),3.86(q,12h),3.03-3.24 (m,18h),2.23-2.31(m,2h),1.96-2.00(m,2h),1.73-1.78(m,4h),1.22(t,18h),0.64(t,4h)。
76.实施例5:
步骤(a):式3-5化合物的制备保持氮气微正压,向20l反应瓶中依次加入4l dmf,149.21 g(1.0 mol)式1-5化合物2-甲基苯并噻唑,2392.2g(10 mol)式2-5化合物1,12-二氯十二烷。加完后,搅匀,反应液升温至150 ℃反应。保温反应6小时,反应完成。
77.反应液冷却至室温,向其中加入3000 ml石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到273.46g(0.704 mol)黄色固体产品。
78.收率70.4%。
79.产品纯度:99.43%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.51(d,1h),8.38(d,1h),7.90(t,1h),7.82(t,1h),4.81-4.89 (m,2h),3.85(t,2h),3.26(s,3h),2.01-2.04(m,2h),1.73-1.79(m,2h),1.46-1.51(m,2h),1.23-1.29(m,14h)。
80.步骤(b):式6-5化合物的制备保持氮气微正压,向50 l反应瓶中依次加入13l四氢呋喃,850.75g(5.0 mol)式4-5化合物4-氯喹啉, 11407.9g(26mol)式5-5化合物1,14-二碘十四烷。加完后,搅匀,反应液升温至66 ℃反应。保温反应75小时,反应完成。
81.反应液冷却至室温,向其中加入10000 ml石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到2179.97g(3.552 mol)黄色固体产品。
82.收率68.3%。
83.产品纯度:98.67%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.49-8.74(m,2h),8.31-8.41(m,2h),δ 8.06-8.21(m,2h), 5.29-5.36(m,2h),2.95(t,2h),2.01-2.04
(m,2h),1.82-1.87(m,2h),1.23-1.29(m,20h)。
84.步骤(c):式7-5化合物的制备保持氮气微正压,向5 l反应瓶中依次加入2.0 l 乙醇,273.46g(0.704 mol)式3-5化合物和2160.58g(3.52 mol)式6-5化合物,搅匀。然后向其中加入35.62g(0.352mol)三乙胺。加完后,搅匀,反应液升温至40 ℃反应。保温反应4小时,反应完成。
85.反应液冷却至室温,向其中加入2l乙酸乙酯和2l石油醚,冷却至0℃,保温过夜,析出大量固体,过滤,收集滤饼得到514.94g(0.615 mol)红色固体产品。
86.收率87.3%。
87.产品纯度:98.94%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.48(d,1h),8.36(d,1h),8.25-8.30(m,3h),7.74-7.93(m,5h),6.87(s,1h),4.81-4.89 (m,2h),3.85(t,2h),3.05-3.21(m,2h),2.95(t,2h),2.01-2.04(m,4h),1.82-1.87(m,2h),1.73-1.79(m,2h),1.46-1.51(m,2h),1.23-1.29(m,34h)。
88.步骤(d):式i-5化合物的制备保持氮气微正压,向20 l反应瓶中依次加入5l 邻二甲苯, 514.94g(0.615 mol)式7-5化合物和637.18g(3.073 mol)(2-10 eq)(n,n-二甲基-3-氨丙基)三甲氧基硅烷,加完后,搅匀;反应液升温至140 ℃反应。保温反应7小时,反应完成。
89.反应液自然冷却至室温,然后继续冷却至0℃,保温析晶3小时,析出大量固体,过滤,收集滤饼得到678.20g(0.541 mol)红色固体产品。
90.收率88.10%,4步反应总收率36.98%,产品纯度:99.28%(hplc);核磁数据:1h nmr (400mhz,dmso-d6):δ 8.49(d,1h),8.37(d,1h),8.25-8.30(m,3h),7.74-7.93(m,5h),6.94(s,1h),4.81-4.89 (m,2h),3.85(t,2h),3.51(bs,18h),3.05-3.21(m,22h),2.01-2.04(m,4h),1.92-1.97(m,4h),1.72-1.77 (m,4h),1.22-1.31(m,36h),0.65(t,4h)。
91.化合物抗菌性能测试:mic (最低抑制浓度) 的测定: 采用微量肉汤稀释法将实施例1-5制得的化合物(i-1,i
ꢀ‑
2,i
ꢀ‑
3,i
ꢀ‑
4,i
ꢀ‑
5)混合在lb 营养肉汤中做系列的二倍稀释,加入定量的受试菌经过一定时间培养后,观察到无细菌生长的最低化合物浓度即为该化合物对此菌的mic (最低抑制浓度)。
92.具体测定步骤如下:( 1) 悬菌液的制备: 在无菌操作台上,用灭菌的接种环挑取适量细菌培养物,移种至10 ml lb 肉汤培养液中,在37 ℃摇床中培养6-8 h,以待菌液至轻微或中度浊度。为保证药敏试验的准确度和精确度,必须对接种菌液的浓度做相应控制。因此,移取少量菌液于比色管中,稀释至0. 5 麦氏标准浓度后稀释1000 倍,菌液含量约为1
×
10
5 cfu /ml。
93.( 2) 抗菌化合物母液的制备: 将化合物溶于无菌水中,制备成特定浓度的抗菌化合物母液,并用无菌过滤头除去溶液可能含有的细菌。
94.( 3) mic 平板的制备: 96 孔板第2 列至第10 列的第2 行至第7 行每孔加入100 μl lb 肉汤,第2 列加100 μl 抗菌化合物母液,用移液枪吹打混匀后吸取100 μl 至第3 列,以此类推,共8 个浓度梯度,第9 列弃去100 μl 的混合液,第10 列不加药液做阳性对照,然后每孔加入100 μl 悬菌液,将混合液用移液枪吹打均匀。第11 列不加菌液加入200 μl lb 肉汤做阴性对照。化合物和菌液吹打混合完毕后,盖上96 孔板盖,置于37 ℃生
化培养箱内培养,培养20-24 h( 大肠杆菌atcc 25922、金黄色葡萄球菌atcc 6538) 或28 ℃生化培养箱内培养40-48 h( 白色念球菌atcc 10231、黑曲霉菌atcc 16404) ,用酶标仪测定菌液的od570值( 光密度) 。
95.( 4) 判定结果: mic (最低抑制浓度)为在96 孔板内完全抑制细菌生长的浓度,具体结果如表1所示。
96.通过上述实验结果可以看出,噻唑橙有机硅季铵盐化合物(化合物i-1、化合物i-2、化合物i-3、化合物i-4、化合物i-5) 对常见菌种(金黄色葡萄球菌atcc 6538,大肠杆菌atcc 25922,白色念球菌atcc 10231,黑曲霉菌atcc 16404)的抑菌效果都很优异,最低抑菌浓度都在35mg/l以下。
97.其中,化合物i-5的抑菌效果尤其优异,对常见菌种的最低抑菌浓度都在3mg/l以下,对黄色葡萄球菌atcc 6538,大肠杆菌atcc 25922和白色念球菌atcc 10231这3种菌种的最低抑菌浓度更是低至0.4mg/l以下。
98.化合物在织物抗菌整理上的性能测试:按照质量百分比计,将实施例1-5制得的化合物(i-1,i-2,i-3,i-4,i-5)料2份和水98份混匀,制得抗菌组合物6-10。
99.将抗菌组合物6-10,浴比1:15,将纯棉织物放入该抗菌整理剂溶液中浸泡10分钟,然后通过压辊,轧余率80%,再将织物放入150℃烘房焙烘5分钟,将织物从烘房取出,分别得到相应的抗菌织物。
100.抗菌织物抗菌性测试: 参考gb/120944.3-2008《纺织品抗菌性能的评价第3部分:振荡法》,菌种选择金黄色葡萄球菌(atcc 6538),大肠杆菌(atcc 25922),白色念球菌(atcc 10231),黑曲霉菌(atcc 16404),具体结果如表2所示。
101.从上述实验结果可以看出,使用本发明的噻唑橙有机硅季铵盐化合物对棉质织物进行抗菌整理后所制得的抗菌织物具有非常优异的抗菌性能,对常见菌种(金黄色葡萄球菌atcc 6538,大肠杆菌atcc 25922,白色念球菌atcc 10231,黑曲霉菌atcc 16404)的抑菌效果都很好,抑菌率均有99.9%。并且,这些抗菌织物都具有良好的耐洗性,都能耐洗涤,100次洗涤后的抑菌率都在90%以上。
102.更优异的是,当化合物分子式中m大于等于8,并小于等于12,n大于等于10,并小于等于15时,耐洗性尤其好,100次洗涤后的抑菌率都保持在99.9%,200次洗涤后的抑菌率都在80%以上,300次洗涤后的抑菌率也都还在70%以上。
103.上述实例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人是能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围;凡根据本发明精神实质所做的等效变换或修饰,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1