钾通道调节剂的制作方法
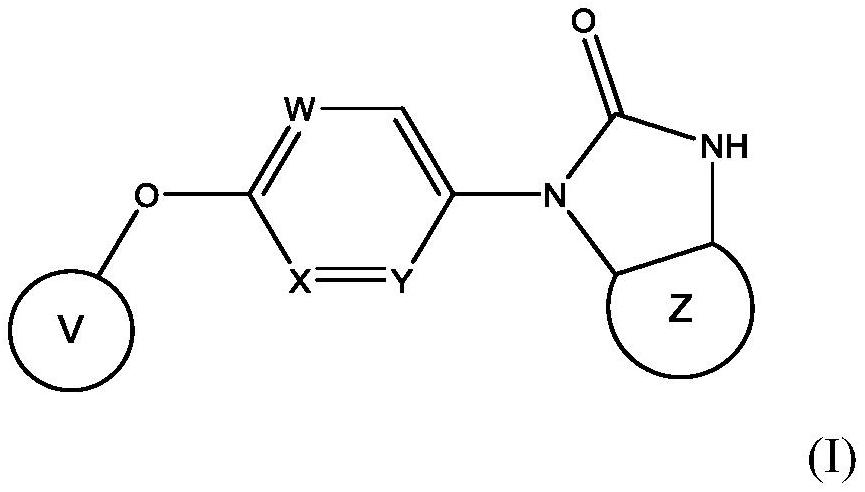
本发明涉及新的化合物、包含它们的药物组合物及其作为药物的用途,特别是在预防或治疗进行性肌阵挛性癫痫(包括与kcnc1基因突变相关的pme)、听力障碍(包括听力损失和耳鸣)以及脆性x染色体综合征、精神分裂症、物质滥用障碍和疼痛中的用途。发明背景kv3电压门控钾通道家族包括四个成员,kv3.1、kv3.2、kv3.3和kv3.4。kv3通道通过将质膜去极化至比-20mv更正的电压而被激活;此外,通道在膜复极化时迅速失活。这些生物物理性质确保通道朝向神经元动作电位的去极化相的峰值打开以启动复极化。由kv3通道介导的动作电位的快速终止允许神经元更快地恢复以达到亚阈值膜电位,从该亚阈值膜电位可以触发进一步的动作电位。作为结果,某些神经元中kv3通道的存在有助于它们在高频下激发的能力(rudy等人,2001)。kv3亚型在cns中占优势,尽管kv3.1、kv3.3和kv3.4通道也在脊髓中发现(brooke等人,2002;2004;2006)。kv3通道亚型由皮质和海马脑区域(例如chow等人,1999;martina等人,1998;mcdonald等人,2006;chang等人,2007)、丘脑(例如kasten等人,2007)、小脑(例如sacco等人,2006;puente等人,2010)和听觉脑干核(li等人,2001)中的中间神经元的亚类所差异表达。对其中一种或多种kv3亚型已被缺失的小鼠的表征表明,kv3.1的缺失导致运动活动增加、脑电图活动改变和片段化睡眠模式(joho等人,1999)。kv3.2的缺失导致癫痫发作阈值的降低和改变的皮层脑电图活动(lau等人,2000)。kv3.3的缺失与轻度共济失调和运动缺陷有关(mcmahon等人,2004)。kv3.1和kv3.3的双缺失产生严重的表型,其特征在于自发性癫痫发作、共济失调和对酒精作用的敏感性增加(espinosa等人,2001;espinosa等人,2008)。人kv3.1基因(kcnc1)中的自发突变引起进行性肌阵挛性癫痫(muona等人,2014)。人kv3.3基因(kcnc3)的突变与脊髓小脑性共济失调(sca13)相关(figueroa等人,2010)。双相情感障碍、精神分裂症是中枢神经系统的严重障碍,其与皮质共生脑回路中的小白蛋白阳性抑制性中间神经元的功能降低相关(reynolds等人,2004;benes等人,2008;brambilla等人,2003;aroniadou-anderjaska等人,2007;ben-ari,2006)。小白蛋白阳性篮状细胞表达kv3通道,允许这些神经元以高频激发,从而在局部回路中提供快速反馈抑制(markram等人,2004)。这种抑制性反馈的准确定时对于维持网络同步是必要的,例如,在与认知功能相关的γ频率场电位振荡的产生中(fisahn等人,2005;engel等人,2001)。在患有精神分裂症的患者中观察到γ振荡的减少(spencer等人,2004),并且有证据启示患有精神分裂症的患者背外侧前额叶皮层中kv3.1的表达减少,但kv3.2的表达没有减少(yanagi等人,2014)。kv3通道的阳性调节剂增强小白蛋白阳性中间神经元的激发(rosato-siri等人,2015;boddum等人,2017),从而导致γ振荡增加(andrade-talavera等人,2020)和在动物模型中挽救认知和社会行为的缺陷(leger等人,2015)。此外,在双相情感障碍小鼠模型中,kv3正调节剂减少了异常行为(parekh等人,2017)。脆性x染色体综合征是具有孤独症特征的儿科发育障碍,其也与小白蛋白阳性中间神经元的功能障碍(例如pirbhoy等人,2020)和kv3.1通道表达的改变(darnell等人,2001;strumbos等人,2010)有关。已显示kv3通道调节剂在脆性x染色体综合征的小鼠模型中在体外和体内挽救听觉脑干功能的缺陷(el-hassar等人,2019)。kv3家族的电压门控离子通道在听觉脑干核中以高水平表达(li等人,2001),其中它们允许将听觉信息从耳蜗传递到较高脑区域的神经元快速激发。认为听觉脑干神经元中kv3.1和kv3.3通道的磷酸化有助于对声音水平的快速生理适应,这可能在暴露于噪声期间起保护作用(desai等人,2008;song等人,2005)。在听力受损小鼠中观察到中枢听觉神经元中kv3.1通道表达的丧失(von hehn等人,2004);此外,kv3.1表达的下降可能与老年小鼠的听力损失有关(jung等人,2005),并且kv3通道功能的损失也可能在噪声创伤诱导的听力损失之后(pilati等人,2012)。此外,听觉脑干网络的病理可塑性可能有助于许多患有不同类型听力损失的人所经历的症状。最近的研究表明,kv3.1通道功能和表达的调节在控制听觉神经元兴奋性中具有主要作用(kaczmarek等人,2005;anderson等人,2018;glait等人,2018;olsen等人,2018;chambers等人,2017),这启示这种机制可以解释一些引起听力相关障碍例如耳鸣的塑性变化。最近,kv3.4通道已成为治疗慢性疼痛的关注靶标。kv3.4通道在背根神经节的神经元上表达(ritter等人,2012;chien等人,2007),其中它们主要在感觉c-纤维上表达(chien等人,2007)。kv3通道也由脊髓中神经元的特定子集表达。特别地,已经在啮齿动物脊髓中鉴定了kv3.1b(deuchars等人,2001;brooke等人,2002)、kv3.3(brooke等人,2006)和kv3.4亚单位(brooke等人,2004),不过,并不总是与涉及感觉处理的回路相关。kv3通道可能形成脊髓神经元(包括运动神经元)的激发特性。此外,最近的研究显示在drg伤害感受器中表达的kv3.4通道对谷氨酸能突触传递具有显著影响(muqeem等人,2018)。动物模型数据表明,在与对疼痛刺激的超敏反应相关的脊髓损伤后,drg神经元中kv3.4通道表面表达下调(ritter等人,2015;zemel等人,2017;zemel等人,2018)。类似地,已经观察到在脊髓结扎后啮齿动物的drg中存在kv3.4表达的下调(chien等人,2007)。后一项研究还表明,给大鼠鞘内施用反义寡核苷酸以抑制kv3.4的表达导致对机械刺激的超敏反应。已经表明,kv3.4通道失活可能受到通道的蛋白激酶c依赖性磷酸化的影响,并且这种生理机制可能允许drg神经元响应于疼痛刺激而改变其激发特征(ritter等人,2012)。这些研究表明机械性异常性疼痛的出现与kv3.4通道表达或功能的降低之间的因果关系。在这些研究的任何一个中都没有评估在sc或drg神经元中kv3.1、kv3.2或kv3.3的表达,并且在drg神经元上没有明确证明这两种亚型的表达(尽管如上所述,它们在脊髓的特定区域内是丰富的)。上述报道的体内研究提供了调节kv3.4作为治疗某些神经性疼痛状态的新方法的基本原理。专利申请wo2011/069951、wo2012/076877、wo2012/168710、wo2013/175215、wo2013/083994、wo2013/182850、wo2017/103604、wo2018/020263、wo2018/109484和wo2020/079422公开了作为kv3.1和kv3.2调节剂的化合物。此外,在癫痫发作、活动过度、睡眠障碍、精神病、听力障碍和双相情感障碍的动物模型中证明了此类化合物的效用。专利申请wo2013/182851公开了某些化合物对kv3.3通道的调节。专利申请wo2013/175211公开了已发现kv3.1、kv3.2和/或kv3.3通道的调节有益于防止或限制由急性噪声暴露引起的永久性听力损失的建立。甚至在停止施用kv3.1、kv3.2和/或kv3.3调节剂之后,也可以观察到此类预防的益处。专利申请wo2017/098254公开了已发现kv3.1、kv3.2和/或kv3.3通道的调节有益于预防或治疗疼痛,特别是神经性疼痛或炎性疼痛。据报道专利申请wo2019/222816、wo20/000065、wo20/089262、wo20/216919和wo2020/216920描述了激活kv3钾通道的化合物。据报道在本技术的优先权日之后公开的专利申请ep3901152和wo2021214090描述了用于治疗认知障碍的kv3增强剂。仍然需要鉴定kv3.1、kv3.2、kv3.3和/或kv3.4的可选择的调节剂,特别是kv3.1和/或kv3.2的调节剂。此类调节剂可以表现出高的体内效力、通道选择性、改善的安全性特征或期望的药代动力学参数,例如高的脑内利用度和/或低清除率,其降低了体内治疗效果所需的剂量。可选择的调节剂可以通过具有不同的来自已知调节剂的代谢物而提供益处。具有平衡的kv3.1、kv3.2、kv3.3和/或kv3.4调节性质的化合物可能是理想的,例如具有相同或相似程度的调节kv3.1和kv3.2的化合物。对于某些治疗适应症,还需要鉴定对kv3.1、kv3.2、kv3.3和/或kv3.4通道具有不同调节作用的化合物,例如,改变通道门控或通道失活的动力学并且可以在体内作为通道的负调节剂起作用的化合物。发明概述本发明提供了式(i)化合物:其中:v为基团(va)、基团(vb)或基团(vc);其中基团(va)和基团(vb)为:其中:r1为h、c1-4烷基、卤素、卤代c1-4烷基、cn、c1-4烷氧基或卤代c1-4烷氧基;r2为h、c1-4烷基、c3-5螺碳环基、卤代c1-4烷基或卤素;r3为h、c1-4烷基、卤代c1-4烷基、卤素;或r3不存在;r13为h、c1-4烷基、卤代c1-4烷基、卤素;或r13不存在;r14为h、c1-4烷基、卤代c1-4烷基、卤素;或r14不存在;a为具有至少一个o原子的5或6元饱和或不饱和杂环;当与苯基一起考虑时,所述杂环任选与环丙基或环丁基或环戊基稠合以形成三环;其中r2和r3可以连接至相同或不同的环原子;r2可以连接至稠合的环原子;并且其中r13和r14可以连接至相同或不同的环原子;其中基团(vc)为:其中:r16为卤素、c1-4烷基、c1-4烷氧基、卤代c1-4烷基、卤代c1-4烷氧基或cn;r17为h、卤素、cn、c1-4烷基、c1-4烷氧基或卤代c1-4烷氧基;r18为h、卤素、cn、c1-4烷基或c1-4烷氧基;w为n或ch;x为n或ch;y为n或ch;其中w、x和y的至少一个为ch,并且当x和y之一为n时,另一个为ch;z为包含一个或两个氮原子的5-元杂芳基,并且其中氮原子之一和碳原子之一可以独立地任选被甲基取代;或z为包含一个或两个氮原子的6-元杂芳基,其中碳原子之一可以任选被甲基取代;并且条件是z不为其中碳原子之一可以任选被甲基取代;或其盐和/或溶剂化物和/或衍生物。式(i)化合物可以以其药学上可接受的盐和/或溶剂化物和/或衍生物的形式提供,例如其盐和/或溶剂化物,特别是其盐。式(i)化合物可以以其药学上可接受的盐和/或溶剂化物的形式提供。在本发明的一个实施方案中,式(i)化合物以药学上可接受的盐的形式提供。式(i)化合物可以用作药物,特别是用于预防或治疗听力障碍,包括听力损失和耳鸣,以及精神分裂症、物质滥用障碍、疼痛或脆性x染色体综合征。此外,提供了用于预防或治疗个体的听力障碍,包括听力损失和耳鸣,以及精神分裂症、物质滥用障碍、疼痛或脆性x染色体综合征的方法,该方法包括施用式(i)化合物。式(i)化合物可以用于制备用于预防或治疗听力障碍,包括听力损失和耳鸣,以及精神分裂症、物质滥用障碍、疼痛或脆性x染色体综合征的药物。式(i)化合物可以用作预防或治疗癫痫,特别是进行性肌阵挛性癫痫,包括与kcnc1基因突变相关的pme的药物。此外,提供了用于预防或治疗个体的癫痫,特别是进行性肌阵挛性癫痫,包括与kcnc1基因突变相关的pme的方法,该方法包括施用式(i)化合物。式(i)化合物可用于制备用于预防或治疗癫痫,特别是进行性肌阵挛性癫痫,包括与kcnc1基因突变相关的pme的药物。还提供了药物组合物,其包含式(i)化合物和药学上可接受的载体或赋形剂。还提供了制备式(i)化合物和用于制备式(i)化合物的新中间体的方法。还提供了式(i)化合物的前药衍生物。发明详述在一个实施方案中,本发明提供了式(i)化合物:其中:v为基团(va)、基团(vb)或基团(vc);其中基团(va)和基团(vb)为:其中:r1为h、c1-4烷基、卤素、卤代c1-4烷基、cn、c1-4烷氧基或卤代c1-4烷氧基;r2为h、c1-4烷基、c3-5螺碳环基、卤代c1-4烷基或卤素;r3为h、c1-4烷基、卤代c1-4烷基、卤素;或r3不存在;r13为h、c1-4烷基、卤代c1-4烷基、卤素;或r13不存在;r14为h、c1-4烷基、卤代c1-4烷基、卤素;或r14不存在;a为具有至少一个o原子的5或6元饱和或不饱和杂环;当与苯基一起考虑时,所述杂环任选与环丙基或环丁基或环戊基稠合以形成三环;其中r2和r3可以连接至相同或不同的环原子;r2可以连接至稠合的环原子;并且其中r13和r14可以连接至相同或不同的环原子;其中基团(vc)为:其中:r16为卤素、c1-4烷基、c1-4烷氧基、卤代c1-4烷基、卤代c1-4烷氧基或cn;r17为h、卤素、cn、c1-4烷基、c1-4烷氧基或卤代c1-4烷氧基;r18为h、卤素、cn、c1-4烷基或c1-4烷氧基;w为n或ch;x为n或ch;y为n或ch;其中w、x和y的至少一个为ch,并且当x和y之一为n时,另一个为ch;z为包含一个或两个氮原子的5-元杂芳基,并且其中氮原子之一和碳原子之一可以独立地任选被甲基取代;或z为包含一个或两个氮原子的6-元杂芳基,其中碳原子之一可以任选被甲基取代;并且条件是z不为其中碳原子之一可以任选被甲基取代。本发明还提供了式(i)化合物的盐。本发明还提供了式(i)化合物的药学上可接受的盐。本发明还提供了式(i)化合物的溶剂化物。本发明还提供了式(i)化合物的药学上可接受的溶剂化物。本发明还提供了式(i)化合物的药学上可接受的盐和/或溶剂化物。本发明还提供了式(i)化合物的药学上可接受的盐和溶剂化物(即药学上可接受的盐的药学上可接受的溶剂化物)。将以下列出的涉及基团(包括v、w、x、y、z、a、r1、r2、r3、r13、r14、r16、r17、r18和z)的相对立体化学和性质的实施方案设想为在适合于环境的情况下(即在化学上敏感的情况下)可彼此独立地完全组合,以形成本发明的另外的实施方案。此类实施方案同样适用于可用于合成式(i)化合物的中间体,例如式(ii)和(iii)、(iv)、(vi)、(ix)和(x)化合物,例如式(ii)和(iii)化合物。在一个实施方案中,v为基团(va)。在第二个实施方案中,v为基团(vb)。基团(va)和(vb)均包含环a并且带有取代基r1、r2、r13和r14。在一个实施方案中,环a为具有至少一个o原子的5元饱和杂环;当与苯基一起考虑时,所述杂环任选与环丙基或环丁基或环戊基稠合以形成三环。环a可以为具有至少一个o原子的5或6元饱和或不饱和杂环,适合地,环a可以为具有至少一个o原子的5元饱和杂环。在一个实施方案中,环a包含一个为氧的杂原子。在一个实施方案中,环a包含两个杂原子,例如两个氧原子或一个氧原子和一个氮原子。在一个实施方案中,环a为二氢呋喃、异噁唑、二氢吡喃、1,3-二氧戊环、1,3-噁嗪或二氢吡喃。适合地,环a为二氢呋喃或二氢吡喃,特别是二氢呋喃。在一个实施方案中,环a选自:其中表示环a与苯环稠合的点。在一个实施方案中,环a选自:其中表示环a与苯环稠合的点。在一个实施方案中,环a选自:其中表示环a与苯环稠合的点,并且“o”和“m”表示与基团a稠合的苯环的邻位和间位。在一个实施方案中,环a选自:其中表示环a与苯环稠合的点,其中“m”和“p”表示与基团a稠合的苯环的间位和对位。适合地,环a为:适合地,环a为:适合地,环a为:特别地,环a为:特别地,环a为:特别地,环a为:在一个实施方案中,当环a为包含一个为氧的杂原子的5元杂环时,其中适合地氧原子位于相对于苯环的酚位置上。在一个实施方案中,r1为h、c1-4烷基、卤素、卤代c1-4烷基或cn,特别是c1-4烷基,例如甲基。在一个实施方案中,r1为h或甲基。在一个实施方案中,r1或h。在一个实施方案中,r1为甲基。在一个实施方案中,当v为基团(vb)时,r1位于对位上,并且为h或甲基:在一个实施方案中,当v为基团(vb)时,r1位于间位上,并且为h或甲基:在一个实施方案中,当v为基团(vb)时,r1位于邻位上,并且为h或甲基:在一个实施方案中,r2为h、c1-4烷基、c3-5螺碳环基或卤素。在一个实施方案中,r2为c1-4烷基,例如甲基或乙基。在一个实施方案中,r2为c3-5螺碳环基,例如c3螺碳环基。在一个实施方案中,r2为甲基。在一个实施方案中,r2为卤素,例如氟。在一个实施方案中,r3为h、c1-4烷基、卤代c1-4烷基或卤素。在一个实施方案中,r3为c1-4烷基,例如甲基。在一个实施方案中,r3为甲基。在另一个实施方案中,r3为卤素,例如氟。在另一个实施方案中,r3不存在。在一个实施方案中,r2和r3位于同一环a原子上。在一个实施方案中,r13为h或不存在。适合地,r13不存在。在一个实施方案中,r14为h或不存在。适合地,r14不存在。v可以选自:在一个实施方案中,v为:在一个实施方案中,v为:在一个实施方案中,v为:在一个实施方案中,v为:基团(vc)带有取代基r16、r17和r18。在一个实施方案中,r16不在对位上。在一个实施方案中,r17和r18之一不为h。在一个实施方案中,r16在间位上。在一个实施方案中,r17在对位上。在一个实施方案中,r16为c1-4烷基、c1-4烷氧基、卤代c1-4烷基、卤代c1-4烷氧基或cn。在一个实施方案中,r16为c1-4烷基、c1-4烷氧基、卤代c1-4烷基或卤代c1-4烷氧基。在一个实施方案中,r16为c1-4烷基、c1-4烷氧基或卤代c1-4烷氧基。在一个实施方案中,r16为卤素、c1-4烷基或c1-4烷氧基。在一个实施方案中,r16为甲基、乙基、丙基、丁基、环丙基、氯、氟、甲氧基、乙氧基、丙氧基、三氟甲基、三氟甲氧基或cn。在一个实施方案中,r16为三氟甲氧基或甲氧基。在一个实施方案中,r16为三氟甲氧基。在一个实施方案中,r16为甲氧基。在一个实施方案中,r17为h、卤素、cn、c1-4烷基或c1-4烷氧基。在一个实施方案中,r17为h、cn、c1-4烷基、c1-4烷氧基或卤代c1-4烷氧基。在一个实施方案中,r17为c1-4烷基或c1-4烷氧基。在一个实施方案中,r17为h、cn或c1-4烷基。在一个实施方案中,r17为h、cn或甲基。在一个实施方案中,r17为甲基、乙基、丙基、丁基、环丙基、氯、氟、甲氧基、乙氧基、丙氧基、三氟甲氧基或cn。在一个实施方案中,r17或h。在一个实施方案中,r17为甲基或cn。在一个实施方案中,r17为甲基。在一个实施方案中,r17为cn。在一个实施方案中,r18为h。在一个实施方案中,r17和r18为h。在一个实施方案中,r16适合地位于邻位或间位上。在该实施方案中,当r16位于邻位上时,其适合地为c1-4烷基,例如甲基、乙基、正丙基、异丙基、环丙基、正丁基或叔丁基。在一个实施方案中,当r16位于间位上时,其适合地为c1-4烷基,例如甲基、乙基、异丙基或环丙基;c1-4烷氧基,例如甲氧基或乙氧基;或卤代c1-4烷氧基,例如三氟甲氧基。在一个实施方案中,r16为位于间位上的三氟甲氧基,并且r17和r18为h。在一个实施方案中,r16为位于间位上的甲氧基,并且r17和r18为h。在一个实施方案中,r18为h,并且r17不为h。在一个实施方案中,r16或r17之一位于邻位上。在该实施方案中,邻位上的取代基适合地为c1-4烷基,例如甲基、乙基、正丙基、异丙基、环丙基、正丁基或叔丁基。在另一个实施方案中,r16和r17之一位于邻位上,而另一个位于间位上。在该实施方案中,邻位上的取代基适合地为c1-4烷基,例如甲基、乙基、正丙基、异丙基、环丙基、正丁基或叔丁基,并且间位上的取代基适合地为c1-4烷基,例如甲基、乙基、异丙基或环丙基;c1-4烷氧基,例如甲氧基或乙氧基;或卤代c1-4烷氧基,例如三氟甲氧基。在一个实施方案中,r16和r17之一位于邻位上,并且另一个位于对位上。在一个实施方案中,r16和r17之一在1-位上,并且另一个在4-位上。在该实施方案中,对位上的取代基适合地为cn、氟或甲基。在一个实施方案中,r16和r17均在邻位上。在该实施方案中,邻位上的取代基适合地为相同的,并且适合地为c1-4烷基,例如甲基、乙基、正丙基、异丙基、环丙基、正丁基或叔丁基。在一个实施方案中,r16在间位上,并且r17在对位上。在此类实施方案中,r16适合地为卤代c1-4烷基,例如三氟甲氧基,并且r17为cn或c1-4烷基,例如cn、甲基、乙基、正丙基、异丙基、环丙基、正丁基或叔丁基,并且特别地r17为cn或甲基。在一个实施方案中,r16为在间位上的三氟甲氧基,并且r17为在对位上的cn。在一个实施方案中,r16为在间位上的三氟甲氧基,并且r17为在对位上的甲基。提及取代基位置编号和命名是关于苯环相对于氧部分的位置进行的,例如:v可以选自:在一个实施方案中,v为:在一个实施方案中,v为:在一个实施方案中,v为:在一个实施方案中,v为:在一个实施方案中,w为n。在一个实施方案中,w为ch。在一个实施方案中,x为n。在一个实施方案中,x为ch。在一个实施方案中,y为n。在一个实施方案中,y为ch。在一个实施方案中,w为n,并且x和y为ch。在一个实施方案中,w为n,x为n,并且y为ch。在一个实施方案中,w为n,x为ch,并且y为n。在一个实施方案中,w和x为ch,并且y为n。在一个实施方案中,基团z为(za):其中:b1、b2、b3和b4各自独立地选自n、ch和c(me);其中b1、b2、b3和b4之一或两个为n,并且仅b1、b2、b3和b4之一可以为c(me);并且其中当b1、b2和b4为ch或c(me)时,b3不为n。在此类实施方案中,表示z与环脲稠合的位置,因此,等同于在一个实施方案中,b1为n。在一个实施方案中,b1为ch。在一个实施方案中,b2为n。在一个实施方案中,b2为ch或c(me)。在一个实施方案中,b2为ch。在一个实施方案中,b2为c(me)。在一个实施方案中,b3为ch或n。在一个实施方案中,b3为n。在一个实施方案中,b3为ch。在一个实施方案中,b3为c(me)。在一个实施方案中,b4为n。在一个实施方案中,b4为ch。在一个实施方案中,b4为c(me)。在一个实施方案中,b1为n,并且b2为c(me)。在一个实施方案中,b1为n,并且b3为c(me)。在一个实施方案中,b1为n,并且b4为c(me)。在一个实施方案中,b1为n,并且b2为n。在一个实施方案中,b1为n,并且b3为n。在一个实施方案中,b1为n,并且b4为n。在一个实施方案中,b1和b3为n,并且b2为ch。在一个实施方案中,b1和b3为n,并且b2为c(me)。在一个实施方案中,b1为n,b2为c(me),b3为n,并且b4为ch。在一个实施方案中,b1为n,并且b2、b3和b4各自独立地为ch。在一个实施方案中,(za)选自:在一个实施方案中,(za)选自:在一个实施方案中,(za)选自:在一个实施方案中,(za)选自:在一个实施方案中,(za)选自:在一个实施方案中,(za)选自:在一个实施方案中,(za)选自:在一个实施方案中,(za)为:在一个实施方案中,(za)为:在一个实施方案中,基团z为基团(zb):其中:c1和c3各自独立地选自ch、c(me)和n;并且c2为nh或n(me);并且其中当c1和c3之一为n时,另一个为ch或c(me)。在一个实施方案中,c1为ch。在一个实施方案中,c1为c(me)。在一个实施方案中,c1为n。在一个实施方案中,c2为nh。在一个实施方案中,c2为n(me)。在一个实施方案中,c3为ch。在一个实施方案中,c3为c(me)。在一个实施方案中,c3为n。在此类实施方案中,表示z与环脲稠合的位置,因此,等同于在一个实施方案中,(zb)选自:在一个实施方案中,(zb)选自:在一个实施方案中,(zb)选自:在一个实施方案中,(zb)选自:在一个实施方案中,(zb)选自:在一个实施方案中,(zb)选自:在一个实施方案中,(zb)为:在一个实施方案中,(zb)为:在一个实施方案中,z为基团(zc):其中:c2为n、ch或c(me),并且c3为ch或c(me);其中当c2或c3之一为c(me)时,另一个为ch。在一个实施方案中,c2为n。在第二个实施方案中,c2为ch或c(me)。在此类实施方案中,表示z与环脲稠合的位置,因此,等同于在一个实施方案中,(zc)选自:在一个实施方案中,z为基团(zd):其中:c1为ch或c(me),并且c2为n、ch或c(me);其中当c1或c2之一为c(me)时,另一个为ch。在一个实施方案中,c2为n。在第二个实施方案中,c2为ch或c(me)。在此类实施方案中,表示z与环脲稠合的位置,其中等同于在一个实施方案中,(zd)选自:在一个实施方案中,z选自(ze-a)、(ze-b)和(ze-c):其中:e1为ch或c(me)。在此类实施方案中,表示z与环脲稠合的位置,因此,分别等同于在一个实施方案中,z为基团(ze-a)。在一个实施方案中,(ze-a)选自:在一个实施方案中,z为基团(ze-b)。在一个实施方案中,(ze-b)选自:在一个实施方案中,z为基团(ze-c)。在一个实施方案中,(ze-c)选自:在一个实施方案中,提供了式(i)化合物,其选自:3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;7-甲基-3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-[6-[4-甲基-3-(三氟甲氧基)苯氧基]-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;7-甲基-3-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-2-吡啶基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-[5-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]吡嗪-2-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[5-[3-(三氟甲氧基)苯氧基]吡嗪-2-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[5-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基吡嗪-2-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[6-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-(2-{2h-螺[1-苯并呋喃-3,1’-环丙烷]氧基}嘧啶-5-基)-1h,2h,3h-咪唑并[4,5-b]吡啶-2-酮;4-[[5-(2-氧代-1h-咪唑并[4,5-b]吡啶-3-基)-2-吡啶基]氧基]-2-(三氟甲氧基)苄腈;7-甲基-3-(2-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基嘧啶-5-基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-[2-(3-甲氧基苯氧基)嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[2-[3-(三氟甲氧基)苯氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;2-甲基-6-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-4h-咪唑并[4,5-c]吡唑-5-酮;2-甲基-6-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-4h-咪唑并[4,5-c]吡唑-5-酮;6-[6-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-3-吡啶基]-2-甲基-4h-咪唑并[4,5-c]吡唑-5-酮;2-甲基-6-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-基)-4h-咪唑并[4,5-c]吡唑-5-酮;2-甲基-6-(2-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基嘧啶-5-基)-4h-咪唑并[4,5-c]吡唑-5-酮;3-[2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-c]吡啶-2-酮;2-甲基-9-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]-7h-嘌呤-8-酮;2-甲基-9-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-7h-嘌呤-8-酮;2-甲基-9-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-7h-嘌呤-8-酮;2-甲基-9-[6-[4-甲基-3-(三氟甲氧基)苯氧基]-3-吡啶基]-7h-嘌呤-8-酮;9-[6-(3-甲氧基苯氧基)-3-吡啶基]-2-甲基-7h-嘌呤-8-酮;9-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-7h-嘌呤-8-酮;9-[6-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-3-吡啶基]-2-甲基-7h-嘌呤-8-酮;3-[2-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[2-[4-甲基-3-(三氟甲氧基)苯氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;6-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-2,4-二氢咪唑并[4,5-c]吡唑-5-酮;3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-c]吡啶-2-酮;1-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-3h-咪唑并[4,5-b]吡啶-2-酮;5-甲基-3-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-基)-1h-咪唑并[4,5-b]吡啶-2-酮;6-甲基-3-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-基)-1h-咪唑并[4,5-b]吡啶-2-酮;和3-[2-[(3,3,7-三甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮。在一个实施方案中,提供了式(i)化合物,其为2-甲基-9-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]-7h-嘌呤-8-酮。在一个实施方案中,提供了式(i)化合物,其为3-[2-[3-(三氟甲氧基)苯氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮。在一个实施方案中,提供了式(i)化合物,其为3-(2-{2h-螺[1-苯并呋喃-3,1’-环丙烷]氧基}嘧啶-5-基)-1h,2h,3h-咪唑并[4,5-b]吡啶-2-酮。在一个实施方案中,提供了式(i)化合物,其为3-[2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮。此类化合物可以以其盐和/或溶剂化物和/或其衍生物(例如其盐和/或溶剂化物)的形式提供。此类化合物还可以以其药学上可接受的盐和/或溶剂化物和/或其衍生物的形式提供,例如其药学上可接受的盐和/或溶剂化物,特别是药学上可接受的盐。适合地,此类化合物不以盐的形式提供。如本文所用的术语“5-元杂芳基”是指包括至少一个杂原子(例如氮)的5元芳族环。5-元杂芳基的实例包括一个氮原子(吡咯)、两个氮原子(咪唑或吡唑)或三个氮原子(三唑)。如本文所用的术语“6-元杂芳基”是指包括至少一个杂原子(例如氮)的6-元芳族环。6-元杂芳基的实例包括一个氮原子(吡啶)或两个氮原子(哒嗪、嘧啶或吡嗪)。如本文所用的术语“卤代”或“卤素”是指氟、氯、溴或碘原子。卤素的特别实例为氟和氯,特别是氟。当化合物含有c1-4烷基时,无论是单独的还是形成较大基团的一部分,例如c1-4烷氧基中,烷基可以为直链、支链、环状或其组合。c1-4烷基的实例为甲基、乙基、正丙基、异丙基、环丙基、正丁基、异丁基、仲丁基、叔丁基和环丁基。提及“丙基”包括正丙基、异丙基和环丙基,并且提及“丁基”包括正丁基、异丁基、仲丁基、叔丁基和环丁基。一组特别的示例性c1-4烷基为甲基、乙基、正丙基、异丙基、环丙基、正丁基、异丁基、仲丁基和叔丁基。c1-4烷氧基的实例包括甲氧基、乙氧基、丙氧基(其包括正丙氧基、异丙氧基和环丙氧基)和丁氧基。术语“c1-4烷氧基”还延伸至其中氧原子位于烷基链内的实施方案,例如-ch2ch2och3或-ch2och3。因此,在一个实施方案中,烷氧基通过碳与分子的其余部分连接。在一个实施方案中,烷氧基通过氧与分子的其余部分连接,例如-oc1-4烷基。本文所用的术语“卤代c1-4烷基”包括被一个或多个卤原子取代的含有1至4个碳原子的直链、支链或环状烷基,例如氟甲基、二氟甲基和三氟甲基。一组特别的示例性卤代c1-4烷基包括被1-3个卤原子,特别是1-3个氟原子取代的甲基和乙基,例如三氟甲基或2,2,2-三氟乙基。本文所用的术语“卤代c1-4烷氧基”包括被一个或多个卤素原子取代的含有1至4个碳原子的直链、支链或环状烷氧基,例如氟甲氧基、二氟甲氧基和三氟甲氧基。一组特别的示例性卤代c1-4烷氧基包括被1-3个卤原子,特别是1-3个氟原子取代的甲氧基和乙氧基。如本文所用的术语“c3-5螺碳环基”指含有3-5个碳原子的环状环系,例如环丙基、环丁基或环戊基,其中环状环系经由螺中心连接至仲碳,使得仲碳是环状环中的3至5个碳原子之一,如下所示:应当理解,为了在药物中使用,式(i)化合物的盐应当是药学上可接受的。适合的药学上可接受的盐对于本领域技术人员来说是显而易见的。药学上可接受的盐包括由berge等人描述的那些。此类药学上可接受的盐包括与无机酸(例如盐酸、氢溴酸、硫酸、硝酸或磷酸)和有机酸(例如琥珀酸、马来酸、乙酸、富马酸、柠檬酸、酒石酸、苯甲酸、对甲苯磺酸、甲磺酸或萘磺酸)形成的酸加成盐。非药学上可接受的盐可以用于例如分离式(i)化合物,并且包括在本发明的范围内。例如,在一个实施方案中,提供了式(i)化合物或其盐。某些式(i)化合物可以与一当量或多当量的酸形成酸加成盐。本发明在其范围内包括所有可能的化学计量和非化学计量形式。式(i)化合物可以以结晶或非结晶形式制备,并且如果是结晶,则可以任选被溶剂化,例如作为水合物。本发明在其范围内包括化学计量的溶剂化物(例如水合物)以及含有可变量溶剂(例如水)的化合物。应当理解,本发明包括式(i)化合物的药学上可接受的衍生物,并且这些衍生物包括在本发明的范围内。如本文所用,“药学上可接受的衍生物”包括式(i)化合物的任何药学上可接受的酯或此类酯的盐,其在施用于接受者时能够提供(直接或间接)式(i)化合物或其活性代谢物或残留物。药学上可接受的衍生物包括药学上可接受的前药。药学上可接受的前药可以通过例如用如下所示例的基团“l”(其中r4和r5如上所述)官能化脲的仲氮来形成:在本发明的一个实施方案中,式(i)化合物通过脲的仲氮被基团l官能化,其中l选自:-po(oh)o-·m+,其中m+为药学上可接受的一价抗衡离子,-po(o-)2·2m+,-po(o-)2·d2+,其中d2+为药学上可接受的二价抗衡离子,-ch(rx)-po(oh)o-·m+,其中rx为氢或c1-3烷基,-ch(rx)-po(o-)2·2m+,-ch(rx)-po(o-)2·d2+,和-co-ch2ch2-co2·m+。应当理解,本发明涵盖式(i)的所有异构体及其药学上可接受的衍生物,包括所有几何、互变异构和光学形式及其混合物(例如外消旋混合物)。当另外的手性中心存在于式(i)化合物中时,本发明的范围内包括所有可能的非对映异构体,包括其混合物。不同的异构体形式可以通过常规方法彼此分离或拆分,或者任何给定的异构体可以通过常规合成方法或通过立体特异性或不对称合成获得。本公开包括本文提供的本发明化合物的所有同位素形式,无论是以下形式:(i)其中给定原子数的所有原子具有在自然界中占优势的质量数(或质量数的混合物)(在本文中称为“天然同位素形式”)或(ii)其中一个或多个原子被具有相同原子数但质量数不同于在自然界中占优势的原子的质量数的原子替代(在本文中称为“非天然变体同位素形式”)。应当理解,原子可以作为质量数的混合物天然存在。术语“非天然变体同位素形式”还包括这样的实施方案,其中具有在自然界中较不常见的质量数的给定原子数的原子的比例(在本文中称为“不常见同位素”)相对于天然存在的原子的比例增加,例如增加至>20%、>50%、>75%、>90%、>95%或>99%的水平(按该原子数的原子的数量计)(后一实施方案称为“同位素富集的变体形式”)。术语“非天然变体同位素形式”还包括其中不常见同位素的比例相对于天然存在的比例已经降低的实施方案。同位素形式可以包括放射性形式(即它们掺入放射性同位素)和非放射性形式。放射性形式典型地为同位素富集的变体形式。因此,化合物的非天然变体同位素形式可以含有一种或多种人工或不常见的同位素,例如在一个或多个原子中的氘(2h或d)、碳-11(11c)、碳-13(13c)、碳-14(14c)、氮-13(13n)、氮-15(15n)、氧-15(15o)、氧-17(17o)、氧-18(18o)、磷-32(32p)、硫-35(35s)、氯-36(36cl)、氯-37(37cl)、氟-18(18f)、碘-123(123i)、碘-125(125i)或与在自然界中在一个或多个原子中占优势的比例相比可以含有增加的比例的所述同位素。包含放射性同位素的非天然变体同位素形式可以例如用于药物和/或底物组织分布研究。放射性同位素氚,即3h和碳-14,即14c鉴于它们易于掺入和现成的检测手段,特别适用于该目的。掺入氘,即2h或d的非天然变体同位素形式,可以提供由更大的代谢稳定性引起的某些治疗优势,例如增加的体内半衰期或减少的剂量需求,因此在一些情况下可能是优选的。此外,可以制备非天然变体同位素形式,其掺入正电子发射同位素,例如11c、18f、15o和13n,并且可用于正电子发射断层扫描(pet)研究以检查底物受体占有率。在一个实施方案中,本发明化合物以天然同位素形式提供。在一个实施方案中,本发明化合物以非天然变体同位素形式提供。在一个特别的实施方案中,非天然变体同位素形式是其中掺入氘(即2h或d)的形式,其中在本发明化合物的一个或多个原子的化学结构中指定氢。在一个实施方案中,本发明化合物的原子为非放射性的同位素形式。在一个实施方案中,本发明化合物的一个或多个原子是放射性的同位素形式。适合地,放射性同位素是稳定的同位素。适合地,非天然变体同位素形式是药学上可接受的形式。在一个实施方案中,提供了本发明化合物,其中化合物的单个原子以非天然变体同位素形式存在。在另一个实施方案中,提供了本发明化合物,其中两个或更多个原子以非天然变体同位素形式存在。非天然同位素变体形式通常可以通过本领域技术人员已知的常规技术或通过本文所述的方法制备,例如类似于所附实施例中描述的用于制备天然同位素形式的方法。因此,非天然同位素变体形式可以通过使用适合的同位素变体(或标记的)试剂替代实施例中使用的常规试剂来制备。由于式(i)化合物旨在用于药物组合物中,因此容易理解,它们各自优选以基本上纯的形式提供,例如至少60%纯,更适合地至少75%纯,并且优选至少85%,特别是至少98%纯(%是基于重量的重量%)。化合物的不纯制备物可以用于制备药物组合物中使用的更纯形式。由于式(i)化合物旨在用于药物组合物中,因此容易理解,它们各自优选以基本上纯的形式提供,例如至少60%纯,更适合地至少75%纯,并且优选至少85%,特别是至少98%纯(%是基于重量的重量%)。化合物的不纯制备物可用于制备药物组合物中使用的更纯的形式。通常,式(i)化合物可以根据本领域技术人员已知的有机合成技术,以及通过下面阐述的代表性方法、实施例中的那些方法及其修饰方法来制备。专利申请wo2011/069951、wo2012/076877、wo2012/168710、wo2013/175215、wo2013/083994、wo2017/098254、wo2017/103604、wo2018/020263、wo2018/109484和wo2020/079422提供了用于合成可用于制备本发明化合物的中间体的方法。一般合成方案以下方案详述了本发明化合物的合成途径和合成此类化合物中的中间体。在以下方案中,反应性基团可以用保护基团保护并且根据本领域技术人员众所周知的已建立的技术脱保护。可以通过下文概括的一般方法制备化合物。在如下描述中,基团v、w、x、y和z具有上述定义的含义,另有描述的除外。方案1a式(i)化合物可以通过环化式(ii)化合物制备,在适合的溶剂例如二氯甲烷中,用羰基化剂例如三光气,优选在相同溶剂中预稀释,并且第二次加入,在0℃在适合的碱例如三乙胺存在下。可选择的是,可以通过使用羰基化剂,例如羰基二咪唑在适合的溶剂例如乙酸乙酯中,在碱例如三乙胺或dipea存在下环化式(ii)化合物制备式(i)化合物。方案1b可以通过金属催化的交叉偶联反应制备式(i)化合物。在该反应中,使式(iii)的芳基卤化物衍生物,其中通常d=cl、br或i,在金属催化剂例如二乙酰氧基钯(乙酸钯(ii)),适合的配体例如5-二苯基磷烷基-9,9-二甲基-呫吨-4-基)-二苯基-磷烷(xantphos)和适合的碱例如碳酸铯存在下,在适合的溶剂例如1,4-二噁烷中,使用常规加热或微波加热反应。方案1c可以通过亲核芳香取代制备式(i)化合物。在该反应中,使式(iv)的芳基卤化物衍生物,其中通常e=f或cl和式(v)的苯酚在适合的碱例如碳酸钾存在下,在适合的溶剂例如n,n-二甲基乙酰胺、n,n-二甲基甲酰胺或二甲亚砜中,采用常规加热或微波加热反应。方案2可以通过还原式(vi)的硝基化合物制备式(ii)的苯胺。将(vi)转化成(ii)的适合的反应条件为例如还原,在fe粉和氯化铵存在下在溶剂例如乙醇/水混合物中,例如在室温或使用常规加热。方案3可以通过以下制备式(iii)的脲:使式(vii)的苯胺和式(viii)的苯胺在适合的溶剂例如二氯甲烷或乙酸乙酯中,与羰基化剂例如三光气,优选在相同溶剂中预稀释,在适合的碱例如三乙胺或二异丙基乙基胺存在下,在0℃-室温的温度范围反应。方案4a可以通过在适合的溶剂例如二氯甲烷中,用羰基化剂例如三光气,在相同溶剂中预稀释并且第二次加入,在0℃在适合的碱例如三乙胺存在下,环化式(ix)化合物制备式(iv)化合物。可选择的是,可以通过使用羰基化剂例如羰基二咪唑在适合的溶剂例如乙酸乙酯中,在碱例如三乙胺或dipea存在下环化式(ii)化合物制备式(i)化合物。方案4b可以通过金属催化的交叉偶联反应制备式(iv)化合物。在该反应中,使式(x)的芳基卤化物衍生物,其中通常d=cl、br或i,在金属催化剂例如二乙酰氧基钯(乙酸钯(ii))、适合的配体例如5-二苯基磷烷基-9,9-二甲基-呫吨-4-基)-二苯基-磷烷(xantphos)和适合的碱例如碳酸铯存在下,在适合的溶剂例如在1,4-二噁烷中,使用常规加热或微波加热反应。方案5可以通过金属催化的交叉偶联反应制备式(vi)化合物。在该反应中,使式(vii)的苯胺和式(xi)的芳基卤化物衍生物,其中通常d=cl、br或i,在金属催化剂例如二乙酰氧基钯(乙酸钯(ii))、适合的配体例如5-二苯基磷烷基-9,9-二甲基-呫吨-4-基)-二苯基-磷烷(xantphos)和适合的碱例如碳酸铯存在下,在适合的溶剂例如在1,4-二噁烷中,使用常规加热或微波加热反应。方案6可以通过还原式(xii)的硝基化合物制备式(vii)的苯胺。将(xii)转化成(vii)的适合的反应条件为例如在fe粉和氯化铵存在下,在溶剂例如乙醇/水混合物中,例如在室温或使用常规加热还原。方案7可以通过还原式(xiii)的硝基化合物制备式(ix)的苯胺。将(xiii)转化成(ix)的适合的反应条件为例如在fe粉和氯化铵存在下,在溶剂例如乙醇/水混合物中,例如在室温或使用常规加热还原。方案8可以通过以下制备式(x)的脲:使式(xiv)的苯胺和式(viii)的苯胺在适合的溶剂例如二氯甲烷或乙酸乙酯中,使用羰基化剂例如三光气,优选在相同溶剂中预稀释,在适合的碱,例如三乙胺或二异丙基乙基胺存在下,在0℃-室温的温度反应。方案9可以通过金属催化的交叉偶联反应制备式(ix)化合物。在该反应中,使式(xiv)的苯胺和式(xi)的芳基卤化物衍生物,其中通常d=cl、br或i在金属催化剂例如二乙酰氧基钯(乙酸钯(ii))、适合的配体例如5-二苯基磷烷基-9,9-二甲基-呫吨-4-基)-二苯基-磷烷(xantphos)和适合的碱例如碳酸铯存在下,在适合的溶剂例如在1,4-二噁烷中,使用常规加热或微波加热反应。方案10可以通过亲核芳香取代制备式(xii)化合物。在该反应中,使式(xv)的芳基卤化物衍生物,其中通常e=f或cl,和式(v)的苯酚在适合的碱例如碳酸钾存在下,在适合的溶剂例如在n,n-二甲基乙酰胺、n,n-二甲基甲酰胺或二甲亚砜中,使用常规加热或微波加热反应。方案11可以通过金属催化的交叉偶联反应制备式(vii)化合物。在该反应中,使式(xiv)的苯胺,其中通常e=br或i,和式(v)的苯酚衍生物在金属催化剂例如碘化亚铜(i)、适合的配体例如吡啶-2-甲酸和适合的碱例如碳酸铯存在下,在适合的溶剂例如在n,n-二甲基乙酰胺中,使用常规加热或微波加热反应。可选择的是,可以通过亲核芳香取代制备式(vii)化合物。在该反应中,式(xiv)的芳基卤化物衍生物,其中通常e=f或cl,和式(v)的苯酚在适合的碱例如碳酸铯存在下,在适合的溶剂例如在二甲亚砜中,使用常规加热或微波加热反应。本发明的方法本发明的另外的方面提供了制备式(i)化合物或其盐,例如其药学上可接受的盐和/或溶剂化物和/或其衍生物的方法,以及制备合成式(i)化合物的中间体或其盐的方法。本发明的方法如上所述并且包括多步方案的任一单个步骤。中间体本发明还涉及合成式(i)化合物的新的中间体。此类新的中间体包括式(ii)、(iii)、(iv)、(vi)、(ix)、(x)和(xiii)化合物。因此,在一个实施方案中,提供了选自以下的化合物:-式(ii)化合物:其中v、w、x、y和z如对式(i)化合物所定义;-式(iii)化合物:其中v、w、x、y和z如对式(i)化合物所定义,并且d为卤素,例如cl、br或i;-式(iv)化合物:其中w、x、y和z如对式(i)化合物所定义,并且e为卤素,例如f或cl;-式(vi)化合物:其中v、w、x、y和z如对式(i)化合物所定义;-式(ix)化合物:其中w、x、y和z如对式(i)化合物所定义,并且e为卤素,例如f或cl;-式(x)化合物:其中w、x、y和z如对式(i)化合物所定义,e为卤素,例如f或cl,并且d为卤素,例如cl、br或i;和-式(xiii)化合物:其中w、x、y和z如对式(i)化合物所定义,并且e为卤素,例如f或cl;本发明还提供了此类中间体的盐,例如药学上可接受的盐。kv3.1、kv3.2、kv3.3和/或kv3.4调节本发明的式(i)化合物为kv3.1的调节剂。式(i)化合物也可以为kv3.2、kv3.3和/或kv3.4的调节剂。可以在生物学实施例1的测定中测试本发明化合物,以确定它们对kv3.1和/或kv3.2和/或kv3.3和/或kv3.4通道的调节特性。如本文所用的“调节剂”是指能够产生由在哺乳动物细胞中重组表达的人kv3.1和/或人kv3.2和/或人kv3.3和/或人kv3.4通道介导的全细胞电流的至少10%增强,并且适合地至少20%增强的化合物。术语“kv3.1、kv3.2、kv3.3和/或kv3.4”应被认为与‘kv3.1和/或kv3.2和/或kv3.3和/或kv3.4’含义相同,并且也可以称作‘kv3.1/kv3.2/kv3.3/kv3.4’。在一个实施方案中,调节剂能够产生由在哺乳动物细胞中重组表达的人kv3.1通道介导的全细胞电流的至少10%增强,并且适合地至少20%增强。适合地,调节剂的pec50在4-8(例如5-7.5)的范围内。在一个实施方案中,调节剂能够产生由在哺乳动物细胞中重组表达的人kv3.2通道介导的全细胞电流的至少10%增强,并且适合地至少20%增强。适合地,调节剂的pec50在4-8(例如5-7.5)的范围内。在一个实施方案中,调节剂能够产生由在哺乳动物细胞中重组表达的人kv3.3通道介导的全细胞电流的至少10%增强,并且适合地至少20%增强。适合地,调节剂的pec50在4-8(例如5-7.5)的范围内。在一个实施方案中,调节剂能够产生由在哺乳动物细胞中重组表达的人kv3.4通道介导的全细胞电流的至少10%增强,并且适合地至少20%增强。适合地,调节剂的pec50在4-8(例如5-7.5)的范围内。在另一个实施方案中,调节剂能够产生由在哺乳动物细胞中重组表达的人kv3.1和kv3.2通道介导的全细胞电流的至少10%的增强,并且适合地至少20%的增强。在另一个实施方案中,调节剂能够产生由在哺乳动物细胞中重组表达的人kv3.1和kv3.3通道介导的全细胞电流的至少10%的增强,并且适合地至少20%的增强。在另一个实施方案中,调节剂能够产生由在哺乳动物细胞中重组表达的人kv3.2和kv3.3通道介导的全细胞电流的至少10%的增强,并且适合地至少20%的增强。在另一个实施方案中,所述调节剂能够产生由在哺乳动物细胞中重组表达的人kv3.1、kv3.2和kv3.3通道介导的全细胞电流的至少10%增强,并且适合地至少20%增强。式(i)化合物或其药学上可接受的盐和/或溶剂化物和/或衍生物可以用于治疗或预防其中需要kv3.1或kv3.2、或kv3.1和kv3.2通道调节剂的疾病或障碍。如本文所用,kv3.1或kv3.2、或kv3.1和kv3.2的调节剂是正或负改变这些通道性质的化合物。在本发明的特别的方面,式(i)化合物是正调节剂。可以在生物学实施例1的测定中测试本发明化合物以确定它们的调节特性。在本发明的一个实施方案中,式(i)化合物或其药学上可接受的盐和/或溶剂化物和/或衍生物对kv3.1通道的调节比对kv3.2通道的调节具有选择性。所谓选择性是指化合物对kv3.1通道的活性是对kv3.2通道的活性的例如至少2倍、5倍或10倍。化合物的活性通过其由ec50值指示的效力适合地定量。在本发明的另一个实施方案中,式(i)化合物或其药学上可接受的盐和/或其溶剂化物和/或其衍生物对kv3.2通道的调节比对kv3.1通道的调节具有选择性。同样,选择性是指化合物对kv3.2通道的活性是对kv3.1通道的活性的例如至少2倍、5倍或10倍。在本发明的特别的实施方案中,式(i)化合物或其药学上可接受的盐和/或其溶剂化物和/或其衍生物显示出在kv3.1和kv3.2通道的调节之间相当的活性,例如一个通道的活性小于另一个通道的活性的2倍,例如小于1.5倍或小于1.2倍。在某些障碍中,使用kv3.3或kv3.1、或kv3.3和kv3.1的调节剂可能是有益的,所述调节剂在两个通道之间显示出特别的选择性分布。例如,化合物对kv3.3通道的调节可以比对kv3.1通道的调节具有选择性,表现出对kv3.3通道的活性是对kv3.1通道的活性的例如至少2倍、5倍或10倍。在本发明的另一个实施方案中,式(i)化合物或其药学上可接受的盐和/或其溶剂化物和/或其衍生物对kv3.1通道的调节比对kv3.3通道的调节具有选择性。同样,所谓选择性是指化合物对kv3.1通道的活性是对kv3.3通道的活性的例如至少2倍、5倍或10倍。在本发明的特别的实施方案中,化合物可以在kv3.3和kv3.1通道的调节之间表现出相当的活性,例如每个通道的活性小于另一个通道的活性的2倍,例如小于1.5倍或小于1.2倍。在某些障碍中,使用kv3.3或kv3.2、或kv3.3和kv3.2的调节剂可能是有益的,所述调节剂在两个通道之间显示出特别的选择性分布。化合物对kv3.3通道的调节相对于对kv3.2通道的调节可以是选择性的,表现出例如对kv3.3通道的活性是对kv3.2通道的活性的至少2倍、5倍或10倍。在本发明的另一个实施方案中,式(i)化合物或其药学上可接受的盐和/或其溶剂化物和/或其衍生物对kv3.2通道的调节比对kv3.3通道的调节具有选择性。同样,所谓选择性是指化合物对kv3.2通道的活性是对kv3.3通道的活性的至少2倍、5倍或10倍。在另一个特别的实施方案中,化合物可以在kv3.3和kv3.2通道的调节之间表现出相当的活性,例如每个通道的活性小于另一个通道的活性的2倍,例如小于1.5倍或小于1.2倍。在本发明的另一个特别的实施方案中,化合物可以在kv3.3、kv3.2和kv3.1通道的调节之间表现出相当的活性,例如每个通道的活性小于任何其它通道的活性的2倍,例如小于1.5倍或小于1.2倍。化合物的活性通过其由ec50值指示的效力适合地定量。治疗方法本发明还提供了式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物,其用于治疗或预防其中需要kv3.1、kv3.2、kv3.3和/或kv3.4调节剂的疾病或障碍,例如下文提及的那些疾病和障碍。本发明提供了治疗或预防其中需要kv3.1、kv3.2、kv3.3和/或kv3.4调节剂的疾病或障碍的方法,例如下文提及的那些疾病和障碍,所述方法包括向个体施用式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物。本发明还提供了式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物在制备用于治疗或预防其中需要kv3.1、kv3.2、kv3.3和/或kv3.4调节剂的疾病或障碍的药物中的用途,所述疾病或障碍例如下文提及的那些疾病和障碍。在一个实施方案中,提供了式(i)化合物或其药学上可接受的盐和/或其溶剂化物和/或其衍生物,其用作药物。本文所用的术语“治疗”包括疾病状态或其症状的控制、缓解、减少或调节。术语“预防”在本文中用于指预防个体的疾病或障碍的症状,或者预防患病个体的疾病或障碍的症状的复发,并且不限于完全预防疾病。个体通常为需要根据本发明治疗或预防的个体。适合地,个体为人。可以通过调节kv3.1和/或kv3.2通道介导的疾病或障碍可以选自以下列表。下面列出的疾病后的括号中的数字是指由美国精神病学协会出版的精神障碍诊断和统计手册第4版(dsm-iv)和/或国际疾病分类第10版(icd-10)中的分类代码。在本发明的一个实施方案中,式(i)化合物或其药学上可接受的盐和/或溶剂化物和/衍生物可以用于治疗或预防选自以下的疾病或障碍:听力障碍、精神分裂症、抑郁和心境障碍、双相情感障碍、物质滥用障碍、焦虑障碍、睡眠障碍、听觉过敏和响度感知障碍、梅尼埃病、平衡障碍和内耳障碍、冲动控制障碍、人格障碍、注意力缺陷/多动障碍、孤独症谱系障碍、进食障碍、认知损伤、共济失调,疼痛例如神经性疼痛、炎性疼痛和混杂的疼痛,路易体痴呆和帕金森病。在本发明的一个实施方案中,式(i)化合物或其药学上可接受的盐和/或溶剂化物和/衍生物可以用于治疗或预防选自以下的疾病或障碍:听力障碍(包括听力损失和耳鸣)、精神分裂症、物质滥用障碍、疼痛(例如神经性疼痛、炎性疼痛和混杂的疼痛)、路易体痴呆和帕金森病。在本发明的一个实施方案中,式(i)化合物或其药学上可接受的盐和/或溶剂化物和/衍生物可以用于治疗或预防选自以下的疾病或障碍:脆性x染色体、雷特障碍和阿尔茨海默病。本发明提供了用于预防或治疗选自以下的疾病或障碍的方法:听力障碍、精神分裂症、抑郁和心境障碍、双相情感障碍、物质滥用障碍、焦虑障碍、睡眠障碍、听觉过敏和响度感知障碍、梅尼埃病、平衡障碍和内耳障碍、冲动控制障碍、人格障碍、注意力缺陷/多动障碍、孤独症谱系障碍、进食障碍、认知损伤、共济失调,疼痛例如神经性疼痛、炎性疼痛和混杂的疼痛,路易体痴呆和帕金森病,该方法包括向个体施用式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或衍生物。本发明还提供了式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物在制备用于治疗或预防选自以下的疾病或障碍的药物中的用途:听力障碍、精神分裂症、抑郁和心境障碍、双相情感障碍、物质滥用障碍、焦虑障碍、睡眠障碍、听觉过敏和响度感知障碍、梅尼埃病、平衡障碍和内耳障碍、冲动控制障碍、人格障碍、注意力缺陷/多动障碍、孤独症谱系障碍、进食障碍、认知损伤、共济失调,疼痛例如神经性疼痛、炎性疼痛和混杂的疼痛,路易体痴呆和帕金森病。在本发明的特别的实施方案中,提供了式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物,其用于治疗或预防听力障碍。听力障碍包括听觉神经病、听觉处理障碍、听力损失,其包括突发性听力损失、噪声诱导的听力损失、物质诱导的听力损失以及60岁以上、65岁以上、70岁以上或75岁以上成年人的听力损失(老年性耳聋)和耳鸣。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防梅尼埃病、平衡障碍和内耳障碍。在本发明的特别的实施方案中,提供了式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物,其用于治疗或预防精神分裂症。精神分裂症包括偏执型(295.30)、紊乱型(295.10)、紧张型(295.20)、未分化型(295.90)和残留型(295.60)亚型;精神分裂症样精神障碍(295.40);情感性分裂症(295.70),包括双相型和抑郁型亚型;妄想症(297.1),包括色情狂型、夸张型、嫉妒型、迫害妄想型、身体型、混合型和未指明型亚型;短时精神障碍(298.8);感应性精神障碍(297.3);由包括具有妄想和幻觉的亚型的一般医学病症引起的精神障碍;物质诱导的精神障碍,包括具有妄想(293.81)和幻觉(293.82)的亚型;和未指明型精神障碍(298.9)。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防抑郁和心境障碍,包括严重抑郁发作、躁狂性发作、混合发作和轻躁狂发作;抑郁障碍,包括严重抑郁障碍、心境恶劣障碍(300.4)、未另外指明的抑郁障碍(311);双相情感障碍,包括i型双相情感障碍、ii型双相情感障碍(伴有轻躁狂发作的复发性严重抑郁发作)(296.89)、循环性精神障碍(301.13)和未另外指明的双相情感障碍(296.80);其它心境障碍,包括由一般医学病症引起的心境障碍(293.83),其包括具有抑郁特征、具有重度抑郁样发作、具有躁狂特征和具有混合特征的亚型)、物质诱导的心境障碍(包括具有抑郁特征、具有躁狂特征和具有混合特征的亚型)和未另外指定的心境障碍(296.90);季节性情感障碍。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防癫痫,(包括但不限于有关定位的癫痫、全身型癫痫、伴有全身性和局限性癫痫发作的癫痫等)、与伦诺克斯-加斯托综合征相关的癫痫发作、作为疾病或障碍的并发症的癫痫发作(例如与脑病、苯丙酮尿症、幼年型戈谢病、lundborg进行性肌阵挛性癫痫、中风、头部损伤、应激、激素变化、药物使用或戒断、酒精使用或戒断、睡眠剥夺、发热、感染等相关的癫痫发作)、特发性震颤、不宁腿综合征、部分性和全身性癫痫发作(包括强直性、阵挛性、强直-阵挛性、失张力性、肌阵挛性、失神型癫痫发作)、继发性全身性癫痫发作、颞叶癫痫、失神性癫痫(包括儿童期、青少年期、肌阵挛性、光诱导和模式诱导的)、严重癫痫性脑病(包括缺氧相关的和rasmussen综合征)、发热惊厥、部分性癫痫持续状态、进行性肌阵挛性癫痫(包括翁-伦病和拉福拉病)、创伤后癫痫发作/癫痫(包括与头部损伤相关的那些)、简单反射性癫痫(包括光敏性、体感性和本体感受性、听源性和前庭性)、通常与癫痫相关的代谢障碍(例如吡哆辛依赖性癫痫)、门凯卷发病、克拉伯病、因酒精和药物滥用(例如可卡因)导致的癫痫发作、与癫痫相关的皮质畸形(例如双皮质综合征或皮质下带状灰质异位),与癫痫发作或癫痫相关的染色体异常,例如部分单体性(15q)/安格尔曼综合征)。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防物质相关性障碍,包括物质使用障碍,例如物质依赖、物质渴求和物质滥用;物质诱导的障碍,例如物质中毒、物质戒断、物质诱导的谵妄、物质诱导的持续性痴呆、物质诱导的持续性遗忘障碍、物质诱导的精神障碍、物质诱导的心境障碍、物质诱导的焦虑障碍、物质诱导的性功能障碍、物质诱导的睡眠障碍和致幻剂持久性知觉障碍(闪回);酒精相关的障碍,如酒精依赖(303.90)、酒精滥用(305.00)、酒精中毒(303.00)、酒精戒断(291.81)、酒精中毒性谵妄、酒精戒断性谵妄、酒精诱导的持续性痴呆、酒精诱导的持续性遗忘障碍、酒精诱导的精神障碍、酒精诱导的心境障碍、酒精诱导的焦虑障碍、酒精诱导的性功能障碍、酒精诱导的睡眠障碍和未另外指明的酒精相关障碍(291.9);安非他明(或安非他明样)相关障碍,例如安非他明依赖(304.40)、安非他明滥用(305.70)、安非他明中毒(292.89)、安非他明戒断(292.0)、安非他明中毒性谵妄、安非他明诱导的精神障碍、安非他明诱导的心境障碍、安非他明诱导的焦虑障碍、安非他明诱导的性功能障碍、安非他明诱导的睡眠障碍和未另外指明的安非他明相关障碍(292.9);咖啡因相关障碍,例如咖啡因中毒(305.90)、咖啡因诱导的焦虑障碍、咖啡因诱导的睡眠障碍和未另外指明的咖啡因相关障碍(292.9);大麻相关障碍,例如大麻依赖(304.30)、大麻滥用(305.20)、大麻中毒(292.89)、大麻中毒性谵妄、大麻诱导的精神障碍、大麻诱导的焦虑障碍和未另外指明的大麻相关障碍(292.9);可卡因相关障碍,例如可卡因依赖(304.20)、可卡因滥用(305.60)、可卡因中毒(292.89)、可卡因戒断(292.0)、可卡因中毒性谵妄、可卡因诱导的精神障碍、可卡因诱导的心境障碍、可卡因诱导的焦虑障碍、可卡因诱导的性功能障碍、可卡因诱导的睡眠障碍和未另外指明的可卡因相关障碍(292.9);致幻剂相关疾病,例如致幻剂依赖(304.50)、致幻剂滥用(305.30)、致幻剂中毒(292.89)、致幻剂持续性感知障碍(闪回)(292.89)、致幻剂中毒性谵妄、致幻剂诱导的精神障碍、致幻剂诱导的心境障碍、致幻剂诱导的焦虑障碍和未另外指明的致幻剂相关疾病(292.9);吸入剂相关障碍,例如吸入剂依赖(304.60)、吸入剂滥用(305.90)、吸入剂中毒(292.89)、吸入剂中毒谵妄、吸入剂诱导的持续性痴呆、吸入剂诱导的精神障碍、吸入剂诱导的心境障碍、吸入剂诱导的焦虑障碍和未另外指明的吸入剂相关障碍(292.9);尼古丁相关障碍,例如尼古丁依赖(305.1)、尼古丁戒断(292.0)和未另外指明的尼古丁相关障碍(292.9);阿片样物质相关障碍,例如阿片样物质依赖(304.00)、阿片样物质滥用(305.50)、阿片样物质中毒(292.89)、阿片样物质戒断(292.0)、阿片样物质中毒性谵妄、阿片样物质诱导的精神障碍、阿片样物质诱导的心境障碍、阿片样物质诱导的性功能障碍、阿片样物质诱导的睡眠障碍和未另外指明的阿片样物质相关障碍(292.9);苯环利定(或苯环利定样)相关障碍,例如苯环利定依赖(304.60)、苯环利定滥用(305.90)、苯环利定中毒(292.89)、苯环利定中毒性谵妄、苯环利定诱导的精神障碍、苯环利定诱导的心境障碍、苯环利定诱导的焦虑障碍和未另外指明的苯环利定相关障碍(292.9);镇静剂-、催眠剂-或抗焦虑剂相关障碍,例如镇静剂、催眠剂或抗焦虑剂依赖(304.10),镇静剂、催眠剂或抗焦虑剂滥用(305.40),镇静剂、催眠剂或抗焦虑剂中毒(292.89),镇静剂、催眠剂或抗焦虑剂戒断(292.0),镇静剂、催眠剂或抗焦虑剂中毒性谵妄,镇静剂、催眠剂或抗焦虑剂戒断谵妄,镇静剂、催眠剂或抗焦虑剂持续性痴呆,镇静剂、催眠剂或抗焦虑剂持续性遗忘障碍,镇静剂、催眠剂或抗焦虑剂诱导的精神障碍,镇静剂、催眠剂或抗焦虑剂诱导的心境障碍,镇静剂、催眠剂或抗焦虑剂诱导的焦虑障碍,镇静剂、催眠剂或抗焦虑剂诱导的性功能障碍,镇静剂,催眠剂或抗焦虑剂诱导的睡眠障碍以及未另外指明的镇静剂、催眠剂或抗焦虑剂相关障碍(292.9);多物质相关障碍,例如多物质依赖(304.80);和其它(或未知)物质相关障碍,例如促蛋白合成类固醇、硝酸盐吸入剂和一氧化二氮。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防焦虑障碍,包括惊恐发作;惊恐障碍,包括惊恐障碍不伴广场恐怖(300.01)和惊恐障碍伴广场恐怖(300.21);广场恐怖;广场恐怖不伴惊恐障碍史(300.22)、特异恐怖症(300.29,以前称为单纯恐怖症),包括动物型、自然环境型、血-注射-损伤型、情境型和其它类型亚型)、社交恐怖症(社交焦虑障碍,300.23)、强迫症(300.3)、创伤后应激障碍(309.81)、急性应激障碍(308.3)、广泛性焦虑障碍(300.02)、由一般医学病症引起的焦虑障碍(293.84)、物质诱导的焦虑障碍、分离焦虑障碍(309.21)、具有焦虑的适应障碍(309.24)和未另外指明的焦虑障碍(300.00)。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防睡眠障碍,包括原发性睡眠障碍,例如睡眠异常,例如原发性失眠(307.42)、原发性睡眠过度(307.44)、发作性睡病(347)、呼吸相关的睡眠障碍(780.59)、昼夜节律睡眠障碍(307.45)和未另外指明的睡眠障碍(307.47);原发性睡眠障碍,例如深眠状态,例如恶梦障碍(307.47)、夜惊症(307.46)、梦游症(307.46)和未另外指明的深眠状态(307.47);与另一种精神障碍相关的睡眠障碍,例如与另一种精神障碍相关的失眠(307.42)和与另一种精神障碍相关的睡眠过度(307.44);由于一般医学病症引起的睡眠障碍,特别是与例如神经障碍、神经性疼痛、不宁腿综合征、心脏和肺部疾病这样疾病相关的睡眠障碍;和物质诱导的睡眠障碍,包括失眠型、睡眠过度型、深眠状态型和混合型亚型;睡眠呼吸暂停和飞行时差反应综合征。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防听觉过敏和响度感知障碍,包括脆性x综合征和孤独症。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防冲动控制障碍,包括:间歇性暴躁障碍(312.34)、偷窃癖(312.32)、病理性赌博(312.31)、纵火癖(312.33)、拔毛癖(312.39)、未另外指明的冲动控制障碍(312.3)、暴食、强迫性购买、强迫性性行为和强迫性囤积。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防性功能障碍,包括性欲障碍,例如性欲过低障碍(302.71)和性厌恶障碍(302.79);性唤起障碍,例如女性性唤起障碍(302.72)和男性勃起障碍(302.72);性欲障碍,例如女性性欲障碍(302.73)、男性性欲障碍(302.74)和早泄(302.75);性交疼痛障碍,例如性交困难(302.76)和阴道痉挛(306.51);未另外指明的性功能障碍(302.70);性欲倒错,例如露阴癖(302.4)、恋物癖(302.81)、摩擦癖(302.89)、恋童癖(302.2)、性受虐癖(302.83)、性虐待癖(302.84)、易装癖(302.3)、窥阴癖(302.82)和未另外指明的性欲倒错(302.9);性身份障碍,例如儿童性身份障碍(302.6)和青少年或成人性身份障碍(302.85);和未另外指明的性障碍(302.9)。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防人格障碍,包括类偏执型人格障碍(301.0)、分裂型人格障碍(301.20)、精神分裂型人格障碍(301.22)、反社会型人格障碍(301.7)、边缘型人格障碍(301,83)、表演型人格障碍(301.50)、自恋型人格障碍(301.81)、回避型人格障碍(301.82)、依赖型人格障碍(301.6)、强迫型人格障碍(301.4)和未另行指明的人格障碍(301.9)亚型。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防注意力缺陷/多动障碍,包括注意力缺陷/多动障碍组合型(314.01)、注意力缺陷/多动障碍-注意障碍为主型(314.00)、注意力缺陷/多动障碍多动-冲动型(314.01)和未另外指明的注意力缺陷/多动障碍(314.9)的亚型;多动障碍;破坏性行为障碍,例如行为障碍,包括儿童期发作型(321.81)、青少年发作型(312.82)和未指明的发作(312.89)的亚型,对立违抗性障碍(313.81)和未另外指明的破坏性行为障碍;和抽动障碍,例如图雷特障碍(307.23)亚型。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防孤独症谱系障碍,包括孤独障碍(299.00)、阿斯伯格障碍(299.80)、雷特障碍(299.80)、童年瓦解性障碍(299.10)和未另外指明的广泛性障碍(299.80,包括非典型孤独症)。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防进食障碍,例如神经性厌食症(307.1),包括限制型和暴食/净化型亚型;神经性贪食症(307.51),包括净化型和非净化型亚型;肥胖症;强迫性进食障碍;暴食症;以及未另外指明的进食障碍(307.50)。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于增强认知,包括治疗其它疾病中的认知损伤,所述其它疾病例如精神分裂症、双相情感障碍、抑郁、其它精神病学障碍和与认知损伤相关的精神病症,例如阿尔茨海默病。可选择的是,式(i)化合物或其药学上可接受的盐和/或溶剂化物可以用于预防认知损伤,例如可能与疾病相关的认知损伤,所述疾病例如精神分裂症、双相情感障碍、抑郁、其它精神病学障碍和与认知损伤相关的精神病症,例如阿尔茨海默病。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防共济失调,包括共济失调,特别是脊髓小脑性共济失调,特别是与r420h、r423h或f448l突变相关的共济失调。式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防疼痛,包括伤害性疼痛、神经性疼痛、炎性疼痛或混杂的疼痛。伤害性疼痛代表对例如皮肤、肌肉、内脏器官、关节、肌腱或骨的组织的有害伤害或损伤的正常反应。构成本发明的一部分的伤害性疼痛的实例包括躯体痛:肌肉骨骼疼痛(关节疼痛、肌盘膜痛)或皮肤疼痛,其通常是良好局限性的;或内脏痛:中空器官或平滑肌。神经性疼痛是由躯体感觉神经系统中的原发病灶或疾病引发或引起的疼痛。感觉异常的范围从感知为感觉异常(麻木)的缺陷到超敏反应(痛觉过敏或异常性疼痛)和感觉迟钝(麻刺感和其它感觉)。构成本发明一部分的神经性疼痛的实例包括但不限于糖尿病性神经病、疱疹后神经痛、脊髓损伤疼痛、幻肢(截肢后)疼痛和中风后中枢性疼痛。神经性疼痛的其它原因包括创伤、化疗和重金属暴露。炎性疼痛是由于在组织炎症部位释放的多种介质激活和致敏伤害性疼痛途径而发生的。在炎性疼痛中作为关键参与者涉及的介质为促炎细胞因子例如il-1-α、il-1-β、il-6和tnf-α、趋化因子、活性氧物质、血管活性胺、脂质、atp、酸和由浸润性白细胞、血管内皮细胞或组织驻留肥大细胞释放的其它因子。构成本发明一部分的炎性疼痛的实例原因包括阑尾炎、类风湿性关节炎、炎性肠病和带状疱疹。混杂的疼痛是指不易分类的疼痛病症或障碍。目前对其潜在机制的理解仍然是初步的,不过,这些障碍的特定疗法是众所周知的;它们包括癌症疼痛、偏头痛和其它原发性头痛以及纤维肌痛类型的广泛疼痛。适合地,可以由kv3.1和/或kv3.2和/或kv3.3和/或kv3.4通道的调节剂介导的特定疼痛适应症为神经性疼痛和/或炎性疼痛。疼痛是主观病症,并且在临床环境中倾向于通过患者的自我评估来测量。因此,可能难以测量和量化痛阈。对于慢性疼痛,通常使用主观11分评定量表,其中0是无疼痛,并且10是可想象的最严重疼痛。个体通常在给定时间段(通常为一天)内记录其最严重的疼痛。还记录最小平均基线评分,并且相对于基线测量对药物的响应,例如,可以观察疼痛从基线评分降低至少10%、20%、30%、40%或50%。由于个体对药物的响应可能不同,因此并非所有个体都可能经历疼痛从基线评分的降低。因此,适合地,在至少10%、20%、30%、40%、50%、60%、70%、80%、90%或所有测试个体中观察到降低。因此,在本发明的一个实施方案中,在将式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物施用于个体时,观察疼痛从基线评分降低至少10%、20%、30%、40%或50%。施用可以发生在预期的疼痛发作之前或疼痛发作之后。在预期疾病或障碍的发展可能导致个体经历的疼痛增加的情况下,可以施用式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物。在个体已经经历疼痛的情况下,可以向个体施用式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物。只要需要治疗,则可以继续治疗个体,例如1天、1周、2周、3周、1个月、6个月、1年、超过1年、超过2年、超过5年或超过10年。因此,在本发明一个实施方案中,将式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物施用于个体达1天至1个月、1周至3个月、1个月至6个月、3个月至1年或超过1年。个体中疼痛的减轻可以通过评估对外部刺激例如机械或热(例如冷)刺激(例如试验部分中所述)的响应来测量。减轻可以被认为是逆转百分比(通过测量受影响的疼痛部位与未受影响的疼痛部位的剂量药前和剂量后阈值来计算,例如在试验部分的数据分析下更详细地描述的)或通过测量受影响的疼痛部位的缩回阈值来计算。优选地,使用百分比逆转计算。因此,在本发明的一个实施方案中,在施用式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物后,对疼痛(例如神经性疼痛或炎性疼痛)的敏感性逆转超过20%、超过30%、超过40%、超过50%、超过60%、超过70%、超过80%或超过90%。适合地,对疼痛的敏感性逆转超过80%或超过90%。个体可以经历后续益处,例如改善的功能、情绪、睡眠、生活质量、减少的工作时间中的一种或多种。在特别的实施方案中,式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防神经性疼痛。在特别的实施方案中,式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防炎性疼痛。在特别的实施方案中,式(i)化合物或其药学上可接受的盐和/或其溶剂化物(例如盐)和/或其衍生物可以用于治疗或预防混杂的疼痛。在一些实施方案中,疼痛为慢性痛。在一个实施方案中,提供了式(i)化合物,用于预防急性噪声诱导的听力损失。在一个实施方案中,提供了预防急性噪声诱导的听力损失的方法,包括向个体施用式(i)化合物。在一个实施方案中,提供了式(i)化合物在制备用于预防急性噪声诱导的听力损失的药物中的用途。急性噪声诱导的听力损失可以由例如暴露于响亮噪声或爆炸的事件引起。在这些情况下,当预期未来事件可能导致急性噪声诱导的听力损失时,可以在该事件之前施用式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物,以预防或减少急性噪声诱导的听力损失。化合物(i)或其药学上可接受的盐、溶剂化物和/或衍生物的施用可以预防任何急性噪声诱导的听力损失,或者可以降低急性噪声诱导的听力损失的严重程度,或者可以减轻由急性噪声诱导的听力损失引起的其它症状,例如耳鸣。将“急性听力损失”定义为历经数小时或数天快速发生的听力损失。例如,听力损失可以历经数分钟、数小时或数天的时间期限发生(例如历经至多1天的时间期限,例如至多2天、3天、4天、5天、6天或7天)。急性听力损失通常由暴露于响亮的声音或爆炸引起。由暴露于响亮的声音或爆炸引起的听力损失在本文中称为“噪声诱导的听力损失”。因此,“急性噪声诱导的听力损失”是指由于暴露于响亮的声音或爆炸引起的历经数小时或数天快速发生的听力损失。急性听力损失的重要症状包括:1.听觉阈值的偏移,即在没有其它声音存在的情况下可以听到的纯音的最小声级的增加;2.耳鸣;以及3.中枢听觉处理的退化,例如听觉时间处理和/或语音理解受损。“响亮的”噪声或爆炸可以是至少90db,例如至少100db、至少110db、至少120db或至少130db。在一个实施方案中,式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物的施用可以在预期引起噪声诱导的急性听力损失的事件之前开始。例如,式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物的施用可以提前至多2周开始,例如在预期会引起噪声诱导的急性听力损失的事件之前至多1周、6天、5天、4天、3天、2天、24小时、12小时、6小时、5小时、4小时、3小时、2小时、1小时、30分钟或至多15分钟开始。式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物可以在预期引起噪声诱导的急性听力损失的事件之前多次施用。在一个实施方案中,式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物在潜在暴露于预期会引起急性噪声诱导的听力损失的噪声或爆炸之前施用,用于预防或减少永久性耳鸣的发展;用于预防或减少听阈的永久偏移的发展;或用于预防或减少永久性退化的中枢听觉处理的发展,包括例如听觉时间处理和/或语音理解。可以理解,预先施用可以是在个体被认为处于暴露于预期会引起急性噪声诱导的听力损失的噪声或爆炸的风险的情况下,并且不限于最终发生这种暴露的那些情况。在一个实施方案中,式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物的施用在预期引起噪声诱导的急性听力损失的事件期间开始。式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物可以在预期会引起噪声诱导的急性听力损失的事件期间多次施用。在一个实施方案中,式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物最初在预期会引起急性噪声诱导的听力损失的噪声或爆炸期间施用,用于预防或减少永久性耳鸣的发展;用于预防或减少听阈的永久偏移的发展;或用于预防或减少永久性退化的中枢听觉处理的发展,包括例如听觉时间处理和/或语音理解。在一个实施方案中,式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物的施用在预期引起急性噪声诱导的听力损失的事件之后开始。因此,在一个实施方案中,式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物最初在预期会引起急性噪声诱导的听力损失的噪声或爆炸之后施用,用于预防或减少永久性耳鸣的发展;用于预防或减少听阈的永久偏移的发展;或用于预防或减少永久性退化的中枢听觉处理的发展,包括例如听觉时间处理和/或语音理解。当式(i)化合物在预期引起急性噪声诱导的听力损失的事件之后施用时,此类施用通常在“急性期”期间进行,即在听力损失已经确定之前进行。在一个实施方案中,式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物的施用可以在预期引起噪声诱导的急性听力损失的事件后至多2个月开始,例如在预期引起急性噪声诱导的听力损失的事件后至多1个月、2周、1周、6天、5天、4天、3天、2天、24小时、12小时、6小时、5小时、4小时、3小时、2小时、1小时、30分钟或至多15分钟开始。式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物可以在预期会引起噪声诱导的急性听力损失的事件之后多次施用。式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物可以历经至多7天(例如,至多1天、至多2天、至多3天、至多4天、至多5天、至多6天或至多7天)、1-2周(例如,7-8天、7-9天、7-10天、7-11天、7-12天、7-13天或7-14天)、2-4周(例如,2-3周或2-4周)或1-2个月(例如,4-6周或4-8周)的期限施用。式(i)化合物或其药学上可接受的盐、溶剂化物和/衍生物最初可以在预期会引起急性噪声诱导的听力损失的噪声或爆炸之前至多1天,例如之前至多2天、之前至多3天、之前至多5天、之前至多1周、之前至多2周或之前至多1个月施用,在暴露于预期会引起急性噪声诱导的听力损失的噪声或爆炸之前的任何点开始的施用通常在暴露于预期会引起急性噪声诱导的听力损失的噪声或爆炸之后持续至多2个月,例如之后至多1个月、之后至多3周、之后至多2周、之后至多1周、之后至多5天、之后至多3天、之后至多2天或之后至多1天。在一个实施方案中,提供了式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物,其用于预防或减少听阈的永久偏移的发展,其中听阈的永久偏移被减少至少10db,例如至少15db、至少20db、至少30db、至少40db或全部。药物组合物为了用于疗法,本发明化合物通常作为药物组合物施用。本发明还提供了药物组合物,其包含式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物和药学上可接受的载体或赋形剂。在一个实施方案中,提供了药物组合物,其包含式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物,其用于治疗或预防选自以下的疾病或障碍:听力障碍、精神分裂症、抑郁和心境障碍、双相情感障碍、物质滥用障碍、焦虑障碍、睡眠障碍、听觉过敏和响度感知障碍、梅尼埃病、平衡障碍和内耳障碍、冲动控制障碍、人格障碍、注意力缺陷/多动障碍、孤独症谱系障碍、进食障碍、认知损伤、共济失调,疼痛例如神经性疼痛、炎性疼痛和混杂的疼痛,路易体痴呆和帕金森病。在另一个实施方案中,提供了用于预防或治疗选自以下的疾病或障碍的方法:听力障碍、精神分裂症、抑郁和心境障碍、双相情感障碍、物质滥用障碍、焦虑障碍、睡眠障碍、听觉过敏和响度感知障碍、梅尼埃病、平衡障碍和内耳障碍、冲动控制障碍、人格障碍、注意力缺陷/多动障碍、孤独症谱系障碍、进食障碍、认知损伤、共济失调,疼痛例如神经性疼痛、炎性疼痛和混杂的疼痛,路易体痴呆和帕金森病,该方法包括向个体施用药物组合物,所述组合物包含式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物。本发明还提供了包含式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物的药物组合物在制备治疗或预防选自以下的疾病或障碍的药物中的用途:听力障碍、精神分裂症、抑郁和心境障碍、双相情感障碍、物质滥用障碍、焦虑障碍、睡眠障碍、听觉过敏和响度感知障碍、梅尼埃病、平衡障碍和内耳障碍、冲动控制障碍、人格障碍、注意力缺陷/多动障碍、孤独症谱系障碍、进食障碍、认知损伤、共济失调,疼痛例如神经性疼痛、炎性疼痛和混杂的疼痛,路易体痴呆和帕金森病。式(i)化合物或其药学上可接受的盐和/或其溶剂化物和/或其衍生物可以通过任意方便的方法施用,例如通过口服、非肠道、口含、舌下、鼻、直肠或透皮施用,并且相应调整药物组合物。其它可能的施用途径包括鼓室内和耳蜗内。当通过口服给予时,可以将具有活性的式(i)化合物或其药学上可接受的盐和/或其溶剂化物和/或其衍生物配制成液体或固体,例如糖浆剂、混悬剂、乳剂、片剂、胶囊剂或锭剂。液体制剂通常由活性成分(例如式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物)在适合的液体载体(例如水性溶剂(例如水、乙醇或甘油)或非水性溶剂(例如聚乙二醇或油))中的混悬剂或溶液剂组成。该制剂还可以包含助悬剂、防腐剂、矫味剂和/或着色剂。片剂形式的组合物可以使用常规用于制备固体制剂的任意适合的药用载体制备,例如硬脂酸镁、淀粉、乳糖、蔗糖和纤维素。胶囊剂形式的组合物可以使用常规包封方法制备,例如包含活性成分(例如式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物)的小丸可以使用标准载体制备,然后填充到硬明胶胶囊中;可选择的是,分散体或混悬液可以使用适合的药用载体制备,例如水性树胶、纤维素、硅酸盐或油,然后将分散体或混悬液填充到软明胶胶囊中。典型的非肠道组合物由活性成分(例如式(i)化合物或其药学上可接受的盐和/或溶剂化物(例如盐)和/或衍生物)在无菌水性载体或非肠道可接受的油(例如聚乙二醇、聚乙烯吡咯烷酮、卵磷脂、花生油或芝麻油)中的溶液剂或混悬剂组成。可选择的是,可以将溶液冻干,然后在即将施用前用适合的溶剂重构。可以将用于经鼻施用的组合物便利地配制成气雾剂、滴剂、凝胶剂和散剂。气雾剂制剂通常包含活性成分在药学上可接受的水性或非水性溶剂中的溶液或精细混悬液,并且通常以无菌形式以单剂量或多剂量存在于密封容器中,所述密封容器可以采取药筒或再填充的形式以与雾化装置一起使用。可选择的是,密封容器可以为一次性分配装置,例如单剂量鼻吸入器或配备计量阀的气雾剂分配器。当剂型包含气雾剂分配器时,其包含抛射剂,所述抛射剂可以为压缩气体,例如空气,或有机抛射剂,例如氟氯烃或氢氟烃。气雾剂剂型也可以采用泵-雾化器的形式。适于口含或舌下施用的组合物包括片剂、锭剂和软锭剂,其中将活性成分与载体例如糖和阿拉伯胶、黄蓍胶或明胶和甘油一起配制。用于直肠施用的组合物方便地为包含常规栓剂基质例如可可脂的栓剂形式。适于透皮施用的组合物包括软膏剂、凝胶剂和贴剂。在一个实施方案中,组合物为单位剂型,例如片剂、胶囊剂或安瓿剂。取决于施用方法的不同,组合物可以包含0.1%-100%重量,例如10-60%重量的活性物质。取决于施用方法的不同,组合物可以包含0%-99%重量,例如40%-90%重量的载体。取决于施用方法的不同,组合物可以包含0.05mg-1000mg,例如1.0mg-500mg的活性物质。取决于施用方法的不同,组合物可以包含50mg-1000mg,例如100mg-400mg的载体。用于治疗上述障碍的化合物的剂量将以通常的方式随障碍的严重程度、患者的体重和其它类似因素的不同而变化。然而,作为一般性指导原则,适合的单位剂量可以为0.05mg-1000mg,更适合地是1.0mg-500mg,并且此类单位剂量可以每天施用多于一次,例如每天两次或三次。此类疗法可以延伸数周或数月。提供给个体的剂量通常为安全有效的剂量,即提供期望的益处与不期望的副作用的可接受的平衡的量。“安全有效量”旨在包括在治疗和/或预防疾病状态中有效实现期望效果的化合物的量。期望的效果通常为例如如下的临床显著的和/或可测量的:(a)预防疾病状态在哺乳动物中发生,特别地,当此类哺乳动物为易患该疾病状态,但尚未被诊断为患有该疾病状态;(b)抑制疾病状态,即减缓或阻止其发展;和/或(c)缓解疾病状态,即引起疾病状态的消退或相关症状的减轻。安全有效量可以是当化合物单独施用时,或者可选择地当其与一种或多种另外的api组合施用时足以实现期望效果的量,所述另外的api是用于本发明的用途的另外的化合物或不同于用于本发明的用途的化合物。为了避免疑问,如本文所述的“安全有效量”可以通过任何适合的剂量方案实现,包括但不限于本文其它部分描述的示例性剂量方案。因此,例如,本文提及施用安全和有效量的化合物,例如通过特别的施用途径,包括通过单剂量或通过多剂量实现安全有效量,例如通过指定的施用途径施用。例如,口服施用安全有效量包括口服施用单剂量和口服施用任意的多个剂量,条件是通过口服施用由此实现安全有效量。在另一个方面,本发明提供了包含式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物(例如包含式(i)化合物或其药学上可接受的衍生物的组合)以及另外的一种或多种药学上可接受的活性成分的组合。本发明提供了式(i)化合物,其用于与另外的一种或多种药学上可接受的活性成分组合使用。当化合物与其它治疗剂组合使用时,化合物可以通过任意便利的途径依次或同时施用。可选择的是,化合物可以单独施用。上述组合可以便利地以药物制剂的形式使用,因此包含如上定义的组合以及药学上可接受的载体或赋形剂的药物制剂构成本发明的另一个方面。此类组合的各个组分可以在分开的或组合的药物制剂中依次或同时施用。组合的各个组分也可以通过相同或不同的途径分开施用。当式(i)化合物或其药学上可接受的衍生物与对相同疾病状态有活性的第二种治疗剂组合使用时,每种化合物的剂量可以与单独使用该化合物时的剂量不同。本领域技术人员将容易理解适合的剂量。适合地,式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物通过口服施用。适合地,式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物以每天2至400mg施用,例如每天2至300mg,特别是每天5至250mg。适合地,将式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物每天施用一次或两次。适合地,将式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物施用至少三个月期限。理想地,式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物通过口服施用,每天一次或两次,每天2-400mg,例如每天2-300mg,特别是每天5-250mg。人个体可以是成人,例如18至65岁。可选择的是,人个体可以是66岁或以上。可以将式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物施用于年龄小于18岁,例如4-17岁的人个体。在进行性肌阵挛性癫痫和脆性x染色体综合征的情况下,对低于18岁的人个体的施用可能具有特别的相关性。为了便利性和有助于患者的依从性,可以使用递送技术例如贴剂或植入剂历经持续的时间期限递送式(i)化合物或其药学上可接受的盐、溶剂化物和/或衍生物,例如至少1周或至少4周。试验本发明通过如下所述的化合物来示例。以下实施例描述了本发明具体化合物的实验室合成,并不意味着以任何方式就化合物或方法而言限制本发明的范围。应当理解,尽管使用了特定的试剂、溶剂、温度和时间期限,但是存在可以用于产生类似结果的许多可能的等效替代方案。本发明意在包括此类等效方案。分析仪器除非另有说明,否则起始材料、试剂和溶剂得自商业供应商,并且无需进一步纯化即可使用。除非另有说明,否则所有具有手性中心的化合物都是外消旋的。当反应被描述为已经以与先前更完整描述的反应类似的方式进行时,所使用的一般反应条件基本上相同。使用的后处理条件具有本领域的标准类型,但可以从一个反应到另一个反应进行调整。起始材料可以不一定已经由所提及的批次制备。合成的化合物可以具有多种纯度,例如85%-99%的范围。在一些情况下,为此调整摩尔数和产率的计算。hplc-质谱(hplc-ms)在与hplc仪器agilent 1100系列偶联的agilent 1100系列lc/msd质谱仪上获取,以正电喷雾电离模式和酸性梯度条件操作。质量控制(3分钟方法):在zorbax sb c18柱(1.8μm 3×50mm)上在酸性条件下进行lc/ms-es+。流动相:a:(h2o+0.05体积%tfa)/b:(ch3cn+0.05体积%tfa)。梯度:t=0分钟0%(b),在2.5分钟内从0至95%(b),95%(b)持续0.2分钟,在0.2分钟内从95至100%(b),100%(b)持续0.4分钟,在0.1分钟内从100%至0%(b)。停止时间4分钟。柱t=60℃。流速:1.5ml/分钟。质量范围es+:(100-1000amu,f=60)。uv检测波长:dad 1a=220.8,dad 1b=254.8。在所述化合物的分析表征中,该方法的应用由“qc_3_min”表示。应当理解,由于例如柱龄这样的因素,在色谱期间测定到的保留时间可以因制备方法的不同而异。手性控制:在od-h(250×4.6mm-5um)上在酸性条件下进行lc/ms-es+。流动相:a:(h2o+0.05体积%tfa)/b:(ch3cn+0.05体积%tfa)。梯度:t=0-6分钟35%(b),t=6-40分钟从35%至50%(b),t=6-40分钟从50%至70%(b),t=45-50分钟从70%至35%(b),t=50-55分钟35%(b)。停止时间60分钟。柱t=40℃。流速:1.0ml/分钟。uv检测波长:dad 1a=220.8,dad 1b=254.8。质子磁共振(nmr)光谱在varian instruments上在300、400、500或600mhz记录,或在bruker instruments上在400mhz记录。使用残留溶剂线作为内标,以ppm(δ)报告化学位移。分裂模式被设计为s(单峰)、br.s(宽单峰)、d(双峰)、t(三重峰)、q(四重峰)、dd(双双峰)、dt(双三重峰)和m(多重峰)。在25-60℃范围的温度记录nmr光谱。在f1和f2两者中使用3355hz的光谱宽度以500ms的混合时间获取2d nmr noesy试验。收集总计256个增量,用线性预测处理至1k,每个扫描8次。在两个维度上用正弦钟形偏移处理数据,并且在f1中用lb=0.3hz处理数据。在许多制备中,使用biotage自动化快速色谱法(sp1和sp4)或flash master personal系统进行纯化。快速色谱在230-400目硅胶(由merck ag darmstadt,germany提供)或300-400目硅胶(由sinopharm chemical reagent co.,ltd.提供)、varian mega be-si预填充柱、预填充biotage硅胶柱(例如biotage snap柱)、预填充的modus硅胶柱上进行。缩写acn 乙腈aq 水性boc 叔丁氧基羰基dcm 二氯甲烷dipea n,n-二异丙基乙基胺etoac 乙酸乙酯g 克h(rs) 小时hatu (o-7-氮杂苯并三唑-1-基)-n,n,n’,n’-四甲基脲鎓六氟磷酸盐)hcl 氯化氢lc/ms 液相色谱-质谱法me 甲基mg 毫克min 分钟ml 毫升mmol 毫摩尔m/z 质荷比nmr 核磁共振mtbe 甲基叔丁基醚rt 室温t 温度t3p 丙膦酸酐tbtu 苯并三唑-1-基-n,n,n’,n’-四甲基脲鎓四氟硼酸盐tea 三乙胺tfa 三氟乙酸vol 体积wt. 重量试验方法中间体12-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-5-硝基-吡啶将2-氯-5-硝基吡啶(60mg,0.38mmol)、3,3-二甲基-2h-苯并呋喃-4-醇(中间体50wo2012/076877,8mg,0.42mmol)和碳酸二钾(碳酸钾)(79mg,0.58mmol)在n,n-二甲基乙酰胺(1.5ml)中的混合物在120℃搅拌1小时。然后用水稀释该混合物,并且用乙酸乙酯(3×20ml)萃取。用盐水(30ml)洗涤合并的有机层,分离,经硫酸钠干燥,过滤,减压浓缩,得到标题化合物2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-5-硝基-吡啶(105mg),为粗棕色半固体。lc/ms:qc_3_min:rt=2.54分钟m/z 287[m+h]+中间体25-硝基-2-[3-(三氟甲氧基)苯氧基]吡啶使用已经对中间体1所述的方法,用3-(三氟甲氧基)苯酚替代3,3-二甲基-2h-苯并呋喃-4-醇(中间体50wo2012/076877)制备中间体2。lc/ms:qc_3_min:rt=2.54分钟m/z 301[m+h]+中间体35-硝基-2-{2h-螺[1-苯并呋喃-3,1’-环丙烷]氧基}嘧啶向2-氯-5-硝基嘧啶(250mg,1.57mmol)在乙腈(2.5ml)中的溶液中加入2h-螺[1-苯并呋喃-3,1’-环丙烷]-4-醇(中间体85wo2012/076877,280mg,1.73mmol)和碳酸钾(325mg,2.36mmol),并且将该混合物在80℃搅拌1小时。用乙酸乙酯(20ml)稀释反应粗物质,并且用盐水(20ml)洗涤。经硫酸钠干燥有机层,过滤,并且蒸发至干,得到标题化合物5-硝基-2-{2h-螺[1-苯并呋喃-3,1’-环丙烷]氧基}嘧啶(430mg),为棕色固体。lc/ms:qc_3_min:rt=2.27分钟m/z 286[m+h]+中间体45-硝基-2-[3-(甲氧基)苯氧基]嘧啶使用已经对中间体3所述的方法,用3-甲氧基苯酚替代2h-螺[1-苯并呋喃-3,1’-环丙烷]-4-醇(中间体85wo2012/076877)制备中间体4。lc/ms:qc_3_min:rt=2.16分钟m/z 248[m+h]+中间体55-硝基-2-[3-(三氟甲氧基)苯氧基]嘧啶将3-(三氟甲氧基)苯酚(2.4562g,13.791mmol)、2-氯-5-硝基嘧啶(2g,12.537mmol)和碳酸二钾(碳酸钾)(2.5991g,18.805mmol)在乙腈(10ml)中的混合物在rt搅拌5小时。用乙酸乙酯(40ml)稀释该混合物,并且用盐水(40ml)洗涤。分离各相,并且经na2so4干燥有机层,过滤,并且蒸发,得到5-硝基-2-[3-(三氟甲氧基)苯氧基]嘧啶(3.1g),不经进一步纯化用于下一步。lc/ms:qc_3_min:rt=2.71分钟m/z 302[m+h]+中间体66-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]吡啶-3-胺向2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-5-硝基-吡啶(中间体1,105mg,0.37mmol)在乙醇(1.6ml)/水(0.4ml)中的混悬液中加入铁(82mg,1.47mmol)和氯化铵(79mg,1.47mmol),并且将该反应混合物在80℃搅拌1小时。过滤出固体,并且真空浓缩滤液。将残留物溶于乙酸乙酯(20ml),并且用盐水(20ml)洗涤。经硫酸钠干燥有机层,过滤,并且蒸发。通过使用sfar 5g作为柱和环己烷/乙酸乙酯80:20-30:70作为洗脱液的硅胶快速色谱(biotage系统)纯化残留物,得到6-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]吡啶-3-胺(60mg),为黄色固体。lc/ms:qc_3_min:rt=1.95分钟m/z 257[m+h]+还可以如对wo2012076877中间体59所述制备中间体6。使用与上述方法类似的方法,用适合的硝基衍生物替代2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-5-硝基-吡啶(中间体1)制备下列化合物。在室温(rt),使用16小时-24小时范围的反应时间进行表中的反应。表中指定了每种情况的条件。通过快速色谱纯化终产物(硅胶柱,使用环己烷/etoac或dcm/甲醇作为洗脱液,或c-18柱,使用水/乙腈作为洗脱液)。中间体10(可选择的路线)6-[3-(三氟甲氧基)苯氧基]嘧啶-3-胺将2-氯-5-氨基嘧啶(48mg,0.3705mmol)、3-(三氟甲氧基)苯酚(103.42mg,0.5807mmol)(0.075ml)、碳酸铯(250mg,0.7673mmol)和二甲亚砜(1ml)的混合物在120℃搅拌16小时。使该反应混合物在饱和nahco3水溶液(30ml)与乙酸乙酯(50ml)之间分配。分离有机层,用盐水(10ml)洗涤,经无水硫酸镁干燥,过滤,然后真空浓缩。通过使用modus12g作为柱和环己烷:乙酸乙酯(0-100%)作为洗脱液的硅胶快速色谱(biotage系统)纯化残留物。合并适合的级分,并且蒸发至干。通过反相色谱法,使用snap c-18 12g柱纯化残留物,用水和乙腈95:5-5:95洗脱。合并适合的级分,蒸发至干,得到2-[3-(三氟甲氧基)苯氧基]嘧啶-5-胺(26mg),为无色油状物。lc/ms:qc_3_min:rt=2.13分钟m/z 272[m+h]+中间体115-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-胺将5-溴吡嗪-2-胺(600mg,3.45mmol)、螺[2h-苯并呋喃-3,1’-环丙烷]-4-醇(中间体85wo2012076877,559mg,3.45mmol)、碳酸铯(2.25g,6.90mmol)、碘化亚铜(i)(131mg,0.69mmol)和吡啶-2-甲酸(254mg,2.07mmol)混合在n,n-二甲基乙酰胺(7ml)中。将该反应混合物分入两个不同的小瓶中,使每个小瓶经历氩气-真空循环,并且在120℃搅拌2小时。用乙酸乙酯稀释该反应混合物(每次20ml),并且经纤维素过滤。用nh4cl饱和溶液(20ml)、盐水(20ml)将滤液洗涤2次,经硫酸钠干燥,并且蒸发至干。通过使用sfar 10g作为柱和环己烷/乙酸乙酯80:20-40:60作为洗脱液的硅胶快速色谱(biotage系统)纯化残留物,得到标题化合物5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-胺(245mg),为棕色粉末。lc/ms:qc_3_min:rt=2.08分钟m/z 256[m+h]+使用与上述方法类似的方法,用适合的苯酚替代螺[2h-苯并呋喃-3,1’-环丙烷]-4-醇(中间体85wo2012076877)制备下列化合物。通过快速色谱纯化终产物(硅胶柱,使用环己烷/etoac或dcm/甲醇作为洗脱液,或c-18柱,使用水/乙腈作为洗脱液)。中间体15n-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-3-硝基-吡啶-2-胺将6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基吡啶-3-胺(wo2012/076877中间体158,50mg,0.19mmol)、碳酸铯(122mg,0.37mmol)和2-氯-3-硝基-吡啶(30mg,0.17mmol)溶于1,4-二噁烷(2ml),并且加入二乙酰氧基钯(乙酸钯(ii))(42mg,0.19mmol)和(5-二苯基磷烷基-9,9-二甲基-呫吨-4-基)-二苯基-磷烷(xantphos)(9mg,0.01mmol)。施加真空-氩气循环3次,然后将该混合物在95℃搅拌2小时。冷却后,用etoac(20ml)稀释该反应,并且用nh4cl饱和溶液(20ml)洗涤。分离两层,并且用盐水(20ml)洗涤有机层,经na2so4干燥,过滤,并且真空蒸发,得到标题化合物n-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-3-硝基-吡啶-2-胺(82mg),为棕色固体。将粗物质不经进一步纯化用于下一步。lc/ms:qc_3_min:rt=2.67分钟m/z 391[m+h]+应用与上述方法类似的方法,用适合的苯胺替代6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基吡啶-3-胺(wo2012/076877中间体158)和用适合的硝基衍生物替代2-氯-3-硝基-吡啶制备下列化合物。还可以使用氮气替代氩气应用该方法。中间体32n2-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]吡啶-2,3-二胺将铁(59mg,1.05mmol)、氯化铵(56mg,1.05mmol)和n-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-3-硝基-吡啶-2-胺(中间体15,82mg,0.21mmol)悬浮于乙醇(4ml)/水(1ml)混合物中。在80℃将该混合物回流1小时。冷却后,过滤出固体,并且用etoac(30ml)稀释滤液,并且用盐水(30ml)洗涤。通过使用sfar 10g作为柱和环己烷/乙酸乙酯80:20-30:70作为洗脱液的硅胶快速色谱(biotage系统)纯化残留物,得到标题化合物n2-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]吡啶-2,3-二胺(42mg),为白色固体。lc/ms:qc_3_min:rt=2.01分钟m/z 361[m+h]+应用与上述方法类似的方法,用适合的硝基衍生物替代n-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-3-硝基-吡啶-2-胺(中间体15)制备下列化合物。用室温(rt)至80℃的温度进行反应。表中指定了每种情况的条件。通过快速色谱(硅胶柱:环己烷/etoac或其它适合的溶剂系统)和/或反相色谱(c-18柱;水/乙腈或其它适合的溶剂系统)纯化终产物。中间体491-(4-氯-2-甲基-嘧啶-5-基)-3-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]脲方法1:将碳酸双(三氯甲基)酯(90mg,0.3033mmol)在二氯甲烷(2ml)中的混合物冷却至0℃。然后滴加6-[3-(三氟甲氧基)苯氧基]吡啶-3-胺(中间体9,200mg,0.7402mmol)和n,n-二异丙基乙基胺(371mg,2.8706mmol)(0.50ml)在二氯甲烷(3ml)中的溶液,并且将该反应混合物保持在0℃搅拌30分钟。施加真空几分钟以除去过量的光气,然后滴加4-(二甲基氨基)吡啶(100mg,0.8185mmol)在二氯甲烷(1ml)中的溶液,并且将该反应混合物保持在0℃搅拌5分钟。加入4-氯-2-甲基-嘧啶-5-胺(115mg,0.8010mmol),并且将该反应混合物保持在0℃搅拌1小时。用dcm(15ml)稀释该反应混合物,并且用0.4m hcl水溶液(10ml)和盐水(15ml)洗涤。分离有机层,经na2so4干燥,过滤,并且真空浓缩。然后通过反相硅胶快速色谱(biotage系统),使用snap c-18 12g柱纯化粗物质,用水:乙腈95:5-5:95洗脱。采集期望的级分,并且真空浓缩,得到标题化合物1-(4-氯-2-甲基-嘧啶-5-基)-3-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]脲(74mg),为白色固体。lc/ms:qc_3_min:rt=2.42分钟;m/z 440&442[m+h]+。使用与上述方法类似的方法,用适合的胺中间体替代6-[3-(三氟甲氧基)苯氧基]吡啶-3-胺(中间体9)制备下列化合物。通过快速色谱(硅胶柱;环己烷/etoac或其它适合的溶剂系统)和/或反相色谱(c-18柱;水/乙腈或其它适合的溶剂系统)纯化终产物。中间体491-(4-氯-2-甲基-嘧啶-5-基)-3-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]脲方法2:将碳酸双(三氯甲基)酯(58mg,0.1955mmol)在乙酸乙酯(3ml)中的混合物冷却至0℃。滴加6-[3-(三氟甲氧基)苯氧基]吡啶-3-胺(中间体9,105mg,0.3886mmol)和n,n-二异丙基乙基胺(148.4mg,1.1483mmol)(0.2ml)在乙酸乙酯(3ml)中的溶液,并且将该反应混合物保持在0℃搅拌15分钟。施加真空几分钟以除去过量的光气,然后加入在乙酸乙酯(0.5ml)和二氯甲烷(0.5ml)中的4-(二甲基氨基)吡啶(48mg,0.3929mmol),并且在0℃搅拌5分钟。加入4-氯-2-甲基-嘧啶-5-胺(62mg,0.4318mmol),并且将该反应混合物保持在室温搅拌1小时。然后用0.2n hcl(20ml)使该反应混合物猝灭,并且用乙酸乙酯(20ml)萃取。经na2so4,干燥有机层,过滤,然后真空浓缩。使用快速柱色谱(biotage系统),应用modus12g柱和环己烷:乙酸乙酯90:10-50:50作为洗脱液,随后进行反相色谱,使用c-18 12g柱和水:乙腈95:5-15:85作为洗脱液纯化残留物,得到标题化合物1-(4-氯-2-甲基-嘧啶-5-基)-3-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]脲(25mg),为白色固体。lc/ms:qc_3_min:rt=2.53分钟;m/z 440&442[m+h]+。使用与上述方法类似的方法,用适合的胺中间体替代6-[3-(三氟甲氧基)苯氧基]吡啶-3-胺(中间体9)制备下列化合物。通过快速色谱(硅胶柱;环己烷/etoac或其它适合的溶剂系统)和/或反相色谱(c-18柱;水/乙腈或其它适合的溶剂系统)纯化终产物。中间体551-(4-氯-2-甲基-嘧啶-5-基)-3-(6-氟-3-吡啶基)脲将碳酸双(三氯甲基)酯(260mg,0.8762mmol)在二氯甲烷(4ml)中的混合物冷却至0℃。滴加5-氨基-2-氟吡啶(190mg,1.6948mmol)和n,n-二异丙基乙基胺(742mg,5.7413mmol)(1ml)在二氯甲烷(4ml)中的溶液,并且将该反应混合物保持在0℃搅拌15分钟。施加真空几分钟以除去过量的光气,然后加入在二氯甲烷(1ml)中的4-(二甲基氨基)吡啶(214mg,1.7517mmol),并且在0℃搅拌5分钟。分批加入在二氯甲烷(2ml)中的4-氯-2-甲基-嘧啶-5-胺(242mg,1.68mmol),并且将该反应混合物保持在室温搅拌1.5小时。然后用0.2n hcl(20ml)使该反应混合物猝灭,并且用乙酸乙酯(20ml)萃取。经na2so4干燥有机层,过滤,然后真空浓缩。通过反相色谱,使用c-18 12g柱和水:乙腈95:5-60:40作为洗脱液纯化残留物,得到标题化合物(分两个批次分离)1-(4-氯-2-甲基-嘧啶-5-基)-3-(6-氟-3-吡啶基)脲(94mg),为黄色固体,和1-(4-氯-2-甲基-嘧啶-5-基)-3-(6-氟-3-吡啶基)脲(180mg),为白色固体。lc/ms:qc_3_min:rt=1.83分钟;m/z 281&283[m+h]+中间体569-(6-氟-3-吡啶基)-2-甲基-7h-嘌呤-8-酮将1-(4-氯-2-甲基-嘧啶-5-基)-3-(6-氟-3-吡啶基)脲(中间体55,94mg,0.3337mmol)、碳酸铯(215mg,0.66mmol)、(5-二苯基磷烷基-9,9-二甲基-呫吨-4-基)-二苯基-磷烷(xantphos)(22mg,0.038mmol)和二乙酰氧基钯(乙酸钯(ii))(7mg,0.0312mmol)在1,4-二噁烷(5ml)中的混合物置于真空-氮气流中,并且在95℃搅拌2小时。过滤该反应,并且真空浓缩,然后通过反相色谱(biotage系统),使用c-18 25g柱和水:乙腈95:5-80:20作为洗脱液纯化,得到标题化合物9-(6-氟-3-吡啶基)-2-甲基-7h-嘌呤-8-酮(12mg),为白色固体。lc/ms:qc_3_min:rt=1.45分钟;m/z 245[m]中间体574-溴-1-[(4-甲氧基苯基)甲基]-3-硝基-吡唑将4-溴-3-硝基-1h-吡唑(135mg,0.70mmol)溶于n,n-二甲基甲酰胺(2ml)。加入氢化钠在矿物油中的60%的分散液(34mg,0.84mmol)和1-(溴甲基)-4-甲氧基-苯(156mg,0.77mmol)。将该反应混合物在rt搅拌2小时。用水(15ml)使反应猝灭,并且用乙酸乙酯(20ml)稀释。分离两相,并且用盐水(20ml)洗涤有机相,经硫酸钠干燥,并且蒸发至干。通过硅胶快速色谱(biotage系统),使用sfar 5g作为柱和环己烷/乙酸乙酯100:0-70:30作为洗脱液纯化残留物,得到标题化合物4-溴-1-[(4-甲氧基苯基)甲基]-3-硝基-吡唑(197mg),为黄色油状物。lc/ms:qc_3_min:rt=2.30分钟;m/z中间体58n-(1-甲基-4-硝基-吡唑-3-基)-6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-吡啶-3-胺将6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡啶-3-胺(wo2012/076877中间体87,394mg,1.55mmol)、碳酸铯(1.01g,3.09mmol)、3-氯-1-甲基-4-硝基吡唑(250mg,1.55mmol)溶于1,4-二噁烷(5ml),加入xantphos(67mg,0.12mmol)和pd(oac)2(17mg,0.08mmol)。施加3次循环氩气-真空,并且将该混合物在110℃搅拌7小时。然后,再加入xanthphos(35mg)和pd(oac)2(10mg),并且将该反应混合物在110℃再搅拌2小时。用nh4cl(50ml)使反应猝灭,并且加入etoac(50ml)。振摇两层,分离,并且采集有机层,用盐水(50ml)洗涤,用na2so4干燥,过滤,并且蒸发。将粗物质悬浮于mtbe(10个体积),并且将该混合物在50℃搅拌1小时,并且在rt搅拌3小时。真空过滤固体,得到n-(1-甲基-4-硝基-吡唑-3-基)-6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-吡啶-3-胺(1g),为棕色固体,不经进一步纯化用于下一步。lc/ms:qc_3_min:rt=2.49分钟;m/z 380[m+h]+。使用与上述方法类似的方法,用适合的胺中间体替代6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡啶-3-胺(wo2012/076877中间体87)制备下列化合物。分离粗物质,或通过快速色谱(硅胶柱;环己烷/etoac或其它适合的溶剂系统)和/或反相色谱(c-18柱;水/乙腈或其它适合的溶剂系统)纯化。中间体63n-[1-[(4-甲氧基苯基)甲基]-4-硝基-吡唑-3-基]-6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-吡啶-3-胺使用与对合成中间体55所述相同的方法,用4-溴-1-[(4-甲氧基苯基)甲基]-3-硝基-吡唑(中间体57)替代3-氯-1-甲基-4-硝基吡唑制备中间体59。lc/ms:qc_3_min:rt=2.71分钟;m/z 486[m+h]+。中间体641-甲基-n3-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)吡唑-3,4-二胺将铁(74mg,1.32mmol)、氯化铵(70mg,1,32mmol)和n-(1-甲基-4-硝基-吡唑-3-基)-6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-吡啶-3-胺(中间体58,100mg,0.26mmol)悬浮在乙醇(2ml)水(0.5000ml)混合物上。在80℃将该混合物回流1小时。冷却后,过滤固体,并且用etoac(30ml)稀释该混合物,并且用盐水(50ml)洗涤。通过硅胶快速色谱(biotage系统),使用sfar 10g作为柱和dcm/meoh 99/1-90/10作为洗脱液纯化残留物,得到标题化合物1-甲基-n3-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)吡唑-3,4-二胺(40mg),为白色固体。lc/ms:qc_3_min:rt=1.92分钟;m/z 350[m+h]+,722[2m+na]+使用与上述方法类似的方法,用适合的硝基衍生物中间体替代n-(1-甲基-4-硝基-吡唑-3-基)-6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-吡啶-3-胺(中间体58)制备下列化合物。用室温(rt)至80℃的温度范围和1小时-64小时的反应时间范围进行反应。表中指定了每种情况的条件。通过快速色谱(硅胶柱;环己烷/etoac或其它适合的溶剂系统)和/或反相色谱(c-18柱;水/乙腈或其它适合的溶剂系统)纯化终产物。中间体702-[(4-甲氧基苯基)甲基]-6-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-4h-咪唑并[4,5-c]吡唑-5-酮向1-[(4-甲氧基苯基)甲基]-n3-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)吡唑-3,4-二胺(中间体69,20mg,0.044mmol)和三乙胺(0.008ml,0.058mmol)在乙酸乙酯(4ml)中的溶液中加入1-1’-羰基二咪唑(10mg,0.064mmol),并且将该反应混合物在室温搅拌2小时。用乙酸乙酯(30ml)稀释该反应,并且用hcl 0.2m溶液(20ml)洗涤。分离两相,并且采集有机相,用盐水(20ml)洗涤,经硫酸钠干燥,过滤,并且蒸发至干。通过硅胶快速色谱(biotage系统),使用sfar 5g作为柱和环己烷/乙酸乙酯60:40-10:90作为洗脱液纯化残留物,得到标题化合物2-[(4-甲氧基苯基)甲基]-6-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-4h-咪唑并[4,5-c]吡唑-5-酮(7mg),为红色油状物。lc/ms:qc_3_min:rt=2.48分钟m/z 482[m+h]+中间体712-氯-n-(3-硝基-2-吡啶基)嘧啶-5-胺向2-氯嘧啶-5-胺(800mg,6.20mmol)在干二噁烷(20ml)的溶液中加入2-氯-3-硝基吡啶(1.96g,12.40mmol)、碳酸铯(4.04g,12.4mmol)、4,5-双(二苯基膦基)-9,9-二甲基呫吨(270mg,0.47mmol)和乙酸钯(ii)(69mg,0.31mmol),并且施加氩气-真空循环3次。将该反应混合物在80℃搅拌1小时。用乙酸乙酯(30ml)稀释该混合物,并且过滤出固体。用水(20ml)和盐水(20ml)洗涤得到的液相,经硫酸钠干燥,过滤,并且蒸发至干,得到标题化合物2-氯-n-(3-硝基-2-吡啶基)嘧啶-5-胺(1.56g),为红棕色固体。lc/ms:qc_3_min:rt=2.09分钟m/z 252[m+h]+中间体72n2-(2-氯嘧啶-5-基)吡啶-2,3-二胺向2-氯-n-(3-硝基吡啶-2-基嘧啶-5-胺(中间体71,1.56g,5.49mmol)在无水乙醇(20ml)和水(5ml)中的溶液中加入氯化铵(588mg,11mmol)和铁粉(615mg,11mmol),并且将该反应混合物在室温搅拌16小时。过滤出固体,并且真空浓缩滤液。将残留物溶于乙酸乙酯(30ml),并且用盐水(50ml)洗涤。经na2so4干燥有机层,过滤,并且蒸发至干。通过硅胶快速色谱(biotage系统),使用sfar 25g作为柱和环己烷/乙酸乙酯80:20至20:80作为洗脱液纯化残留物,得到标题化合物n2-(2-氯嘧啶-5-基)吡啶-2,3-二胺(800mg),为棕色固体。lc/ms:qc_3_min:rt=1.29分钟m/z 222[m+h]+中间体733-(2-氯嘧啶-5-基)-1h-咪唑并[4,5-b]吡啶-2-酮向n2-(2-氯嘧啶-5-基)吡啶-2,3-二胺(中间体72,800mg,3.62mmol)在二氯甲烷(20ml)中的溶液中加入三乙胺(1.32ml,7.24mmol),并且将该混合物冷却至0℃。滴加作为二氯甲烷(15ml)溶液的三光气(429mg,1.45mmol)。将该混合物在0℃搅拌10分钟。用二氯甲烷(30ml)稀释该混合物,并且用hcl 0.2m溶液(20ml)和盐水(5ml)洗涤。经硫酸钠干燥有机层,过滤,并且蒸发至干,得到不溶性固体,将其悬浮于乙酸乙酯(15ml),并且搅拌过夜。过滤后,采集固体,并且干燥,得到标题化合物3-(2-氯嘧啶-5-基)-1h-咪唑并[4,5-b]吡啶-2-酮(140mg),为浅棕色固体。lc/ms:qc_3_min:rt=1.75分钟m/z 248[m+h]+中间体742-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-n-(4-硝基-3-吡啶基)嘧啶-5-胺将2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-胺(wo2012/076877中间体65,50mg,0.19mmol)、3-溴-4-硝基-吡啶(43mg,0,21mmol)、xantphos(17mg,0.03mmol)、碳酸铯(127mg,0.39mmol)和pd(oac)2(4mg,0.02mmol)在1,4-二噁烷(2ml)中的混合物置于真空-氮气流中,并且在95℃搅拌1.5小时。然后用etoac(15ml)稀释该混合物,并且用水(20ml),然后用盐水(20ml)洗涤。分离有机层,经na2so4干燥,过滤,并且真空浓缩,得到标题化合物2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-n-(4-硝基-3-吡啶基)嘧啶-5-胺(104mg),不经进一步纯化用于下一步。lc/ms:qc_3_min:rt=2.47分钟m/z 380[m+h]+使用与上述方法类似的方法,用适合的苯胺替代2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-胺(wo2012/076877中间体65)和用适合的硝基衍生物替代3-溴-4-硝基-吡啶制备下列化合物。在95℃将反应加热1.5-2小时。分离粗物质,或通过快速色谱(硅胶柱;环己烷/etoac或其它适合的溶剂系统)和/或反相色谱(c-18柱;水/乙腈或其它适合的溶剂系统)纯化产物。中间体79n3-[2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]吡啶-3,4-二胺将2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-n-(4-硝基-3-吡啶基)嘧啶-5-胺(中间体74,104mg,0.27mmol)、铁(61mg,1.1mmol)和氯化铵(59mg,1.1mmol)在乙醇(4ml)和水(1ml)中的混合物在rt搅拌16小时。过滤该反应混合物,并且用etoac(10ml)洗涤固体。采集有机滤液,并且用水(10ml)、然后用盐水(10ml)洗涤。分离有机层,经na2so4干燥,过滤,并且真空浓缩。然后通过反相快速色谱(biotage系统),使用c-18固定相,应用snap c-18 12g柱纯化粗物质,用水:乙腈95:5-0:100洗脱。采集期望的级分,并且真空浓缩,得到标题化合物n3-[2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]吡啶-3,4-二胺(23mg),为白色固体。lc/ms:qc_3_min:rt=2.07分钟m/z 350[m+h]+使用与上述方法类似的方法,用适合的硝基衍生物替代2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-n-(4-硝基-3-吡啶基)嘧啶-5-胺(中间体72)制备下列化合物。用室温(rt)至90℃的温度范围进行反应。表中指定了每种情况的条件。通过快速色谱(硅胶柱;环己烷/etoac或其它适合的溶剂系统)和/或反相色谱(c-18柱;水/乙腈或其它适合的溶剂系统)纯化终产物。中间体841-(2-溴-3-吡啶基)-3-[2-[3-(三氟甲氧基)苯氧基]嘧啶-5-基]脲将碳酸双(三氯甲基)酯(32mg,0.1078mmol)在二氯甲烷(1.5ml)中的混合物冷却至0℃。然后滴加2-[3-(三氟甲氧基)苯氧基]嘧啶-5-胺(中间体10,75mg,0.2765mmol)和n,n-二异丙基乙基胺(148mg,1.1452mmol)(0.20ml)在二氯甲烷(1.5ml)中的溶液,并且将该反应混合物保持在0℃搅拌1小时。施加真空几分钟以除去过量的光气,然后滴加4-(二甲基氨基)吡啶(35mg,0.2865mmol)在二氯甲烷(0.5000ml)中的溶液,并且将该反应混合物保持在0℃搅拌5分钟。加入2-溴吡啶-3-胺(55mg,0.3179mmol),并且将该反应混合物保持在0℃搅拌1小时。真空浓缩该混合物,然后通过反相硅胶快速色谱(biotage系统),使用snap c-18 12g柱纯化粗物质,用水:乙腈95:5至30:70洗脱。采集期望的级分,并且真空浓缩,得到1-(2-溴-3-吡啶基)-3-[2-[3-(三氟甲氧基)苯氧基]嘧啶-5-基]脲(18mg),为白色固体。lc/ms:qc_3_min:rt=2.04分钟m/z 470&472[m+h]+实施例13-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮向n2-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]吡啶-2,3-二胺(中间体32,21mg,0.06mmol)(中间体29)在etoac(4ml)中的溶液中加入1-1’-羰基二咪唑(10.4mg,0.06mmol),并且将该反应混合物在室温搅拌2小时。用etoac(30ml)稀释该反应,并且用hcl 0.2m溶液(20ml)洗涤。分离两相,并且采集有机相,用盐水(20ml)洗涤,经硫酸钠干燥,过滤,并且蒸发至干。通过c-18快速色谱,使用sfar 10g作为柱和h2o/acn 95:5-20:80作为洗脱液纯化残留物。采集期望的级分,并且真空除去溶剂,得到标题化合物3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮(17mg,为白色固体。lc/ms:qc_3_min:rt=2.32分钟m/z 387[m+h]+1h-nmr(400mhz;dmso-d6):δppm 11.44(bs,1h),8.37(d,1h),8.07(dd,1h),7.90(dd,1h),7.35-7.41(m,1h),7.05-7.11(m,2h),6.90(d,1h),6.43(d,1h),4.42(s,2h),2.11(s,3h),1.07-1.20(m,2h),0.83-0.88(m,2h)。实施例27-甲基-3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮将4-甲基-n2-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]吡啶-2,3-二胺(中间体33,25mg,0.07mmol)和三乙胺(20ul,0.17mmol)混合在二氯甲烷(5ml)中,并且将混合物冷却至0℃。缓慢地加入三光气(8mg,0.03mmol)在二氯甲烷(2ml)中的溶液,并且将该反应混合物在0℃搅拌30分钟。用乙酸乙酯(30ml)稀释该反应,并且用0.2m hcl水溶液(20ml)洗涤。分离两相,并且用盐水(20ml)洗涤有机相,经硫酸钠干燥,过滤,并且蒸发至干。通过硅胶快速色谱(biotage系统),使用sfar 5g作为柱和dcm/meoh99.5:0.5至95:5作为洗脱液纯化残留物。采集期望的级分,并且真空除去溶剂,得到标题化合物7-甲基-3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮(9mg)(实施例2),为白色固体。lc/ms:qc_3_min:rt=2.39分钟m/z 401[m+h]+401&823[2m+na]+。使用与上述方法类似的方法,用适合的二胺替代4-甲基-n2-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]吡啶-2,3-二胺(中间体33)制备下列化合物。通过快速色谱(硅胶柱,使用环己烷/etoac或dcm/甲醇作为洗脱液,和/或c-18柱,使用水/乙腈作为洗脱液)纯化终产物。实施例173-[2-[3-(三氟甲氧基)苯氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮向n2-[2-[3-(三氟甲氧基)苯氧基]嘧啶-5-基]吡啶-2,3-二胺(中间体48,786mg,2.1636mmol)在乙酸乙酯(5ml)中的溶液中加入1-1’-羰基二咪唑(491.15mg,3.029mmol)和三乙胺(580.4mg,5.7357mmol),将该反应混合物在室温搅拌4小时。用乙酸乙酯(50ml)稀释该反应,并且用0.2n hcl水溶液(30ml)洗涤。分离两相,并且采集有机相,用盐水(30ml)洗涤,经硫酸钠干燥,过滤,并且蒸发至干。将残留物混悬于iproh(10个体积),并且将该混合物在80℃搅拌至完全溶解,然后在rt搅拌过夜。真空过滤出固体,并且真空蒸发滤液,溶于etoac,并且用sfar硅胶柱10g过滤。将该滤液与在先的固体放置在一起,并且将粗物质混悬于etoac(10个体积)。将该混合物在70℃搅拌至完全溶解,然后加入环己烷(10个体积)。关闭加热,并且将该混合物在rt搅拌2小时。真空过滤固体,得到3-[2-[3-(三氟甲氧基)苯氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮(424mg),为淡粉色固体。lc/ms:qc_3_min:rt=2.63分钟;m/z 390[m+h]+1h-nmr(400mhz;dmso-d6):δppm 11.56(s,1h),8.98(s,2h),7.93(dd,1h),7.58(t,1h),7.40-7.44(m,2h),7.35(ddd,1h),7.26-7.32(m,1h),7.11(dd,1h)实施例242-甲基-9-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]-7h-嘌呤-8-酮将1-(4-氯-2-甲基-嘧啶-5-基)-3-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]脲(中间体49,27mg,0.0614mmol)、碳酸铯(140mg,0.4297mmol)、xantphos(24mg,0.0415mmol)和pd(oac)2(5mg,0.0223mmol)在1,4-二噁烷(1ml)中的混合物置于真空-氮气流中并且在95℃搅拌2小时。用nh4cl(10ml)和水(10ml)使该反应混合物猝灭,然后用乙酸乙酯(15ml)萃取。用盐水(15ml)洗涤有机层,经na2so4干燥,过滤,然后真空浓缩。快速色谱(biotage系统)用于纯化残留物,使用modus 5g柱和环己烷:乙酸乙酯90:10-0:100、随后dcm:甲醇80:20。真空浓缩级分,再通过反相色谱,使用c-18 12g柱和水:乙腈95:5-40:60作为洗脱液纯化,得到标题化合物2-甲基-9-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]-7h-嘌呤-8-酮(1mg),为白色固体。lc/ms:qc_3_min:rt=2.19分钟;m/z 404[m+h]+1h-nmr(400mhz;cdcl3):δppm 9.11(s,1h),8.58(dd,1h),8.30(s,1h),8.10(dd,1h),7.42-7.47(m,1h),7.13-7.18(m,2h),7.09-7.13(m,2h),2.66(s,3h)。使用与上述方法类似的方法,用适合的脲中间体替代1-(4-氯-2-甲基-嘧啶-5-基)-3-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]脲制备下列化合物。通过快速色谱(硅胶柱;环己烷/etoac或其它适合的溶剂系统)和/或反相色谱(c-18柱;水/乙腈或其它适合的溶剂系统)纯化终产物。实施例309-[6-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-3-吡啶基]-2-甲基-7h-嘌呤-8-酮将9-(6-氟-3-吡啶基)-2-甲基-7h-嘌呤-8-酮(中间体56,12mg,0.0489mmol)、碳酸二钾(11mg,0.0796mmol)和3,3-二甲基-2h-苯并呋喃-4-醇(中间体50wo2012/076877,8.5mg,0.0518mmol)在二甲亚砜(5ml)中的混合物置于真空-氮气流中,并且在120℃搅拌1小时,然后在135℃搅拌4小时。加入3-3-二甲基-2h-苯并呋喃-4-醇(2mg),并且将该反应混合物在135℃搅拌4小时。过滤该反应,并且真空浓缩。然后用乙酸乙酯(20ml)稀释残留物,然后用水(5×20ml)洗涤。然后经na2so4干燥有机层,然后真空浓缩。在单独的小瓶中,将9-(6-氟-3-吡啶基)-2-甲基-7h-嘌呤-8-酮(中间体56,17mg,0.0693mmol)、碳酸二钾(碳酸钾)(14mg,0.1013mmol)、3,3-二甲基-2h-苯并呋喃-4-醇(12mg,0.0731mmol)在二甲亚砜(1ml)中的混合物置于真空-氮气流中,并且在120℃搅拌1小时,然后在135℃搅拌4小时。加入3-3-二甲基-2h-苯并呋喃-4-醇(2mg),并且将该反应混合物在135℃搅拌24小时。用水(10ml)使该反应混合物猝灭,然后用乙酸乙酯(2×10ml)萃取。然后用水(2×10ml)、然后用盐水(10ml)洗涤有机层,经na2so4干燥,过滤,然后真空浓缩。此时,合并两个反应小瓶用于纯化。然后通过快速柱色谱(biotage系统),使用modus12g柱和dcm:甲醇99:1-95:5作为洗脱液,随后通过反相色谱(biotage系统),使用c-18 25g柱和水:乙腈95:5-55:45纯化残留物。分离标题化合物9-[6-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-3-吡啶基]-2-甲基-7h-嘌呤-8-酮(1.2mg),为白色固体。lc/ms:qc_3_min:rt=2.07分钟;m/z 390[m+h]+实施例313-[2-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮向3-(2-氯嘧啶-5-基)-1h-咪唑并[4,5-b]吡啶-2-酮(中间体73,15mg,0.061mmol)在n,n-二甲基甲酰胺(1ml)中的溶液中加入7-甲基-2h-螺[1-苯并呋喃-3,1’-环丙烷]-4-醇(中间体156wo2012/076877,12mg,0.067mmol)和碳酸钾(13mg,0.092mmol),并且将该混合物在90℃搅拌20小时。用乙酸乙酯(10ml)稀释该混合物,并且用水(10ml)和盐水(10ml)洗涤。经硫酸钠干燥有机层,过滤,并且蒸发至干。通过硅胶快速色谱(biotage系统),使用sfar 10g作为柱和环己烷/乙酸乙酯60:40-0:100作为洗脱液纯化残留物。再通过使用c18固定相的反相快速色谱(biotage系统),应用snap 12g作为柱和水/乙腈70:30-10:90作为洗脱液纯化得到的残留物,得到标题化合物3-[2-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮(3.80mg),为白色固体。lc/ms:qc_3_min:rt=2.26m/z 388[m+h]+使用与上述方法类似的方法,用适合的苯酚替代7-甲基-2h-螺[1-苯并呋喃-3,1’-环丙烷]-4-醇(中间体156wo2012/076877)制备下列化合物。通过快速色谱(硅胶柱,使用环己烷/etoac或dcm/甲醇作为洗脱液,或c-18柱,使用水/乙腈作为洗脱液)纯化终产物。实施例346-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-2,4-二氢咪唑并[4,5-c]吡唑-5-酮将2-[(4-甲氧基苯基)甲基]-6-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-4h-咪唑并[4,5-c]吡唑-5-酮(中间体70,8mg,0.017mmol)溶于三氟乙酸(1ml,10mmol),并且将该反应混合物在70℃搅拌5天。蒸发溶剂,并且通过使用c18固定相的反相快速色谱(biotage系统),应用sfar 12g作为柱和水/乙腈80:20-75:25作为洗脱液纯化残留物,得到标题化合物6-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-2,4-二氢咪唑并[4,5-c]吡唑-5-酮(1.2mg),为白色固体。lc/ms:qc_3_min:rt=2.07m/z 362[m+h]+实施例353-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-c]吡啶-2-酮将n3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]吡啶-3,4-二胺(中间体80,30mg,0.0832mmol)和n,n-二异丙基乙基胺(22.26mg,0.1722mmol)(0.03ml)在二氯甲烷(1ml)中的混合物冷却至0℃。滴加碳酸双(三氯甲基)酯(10mg,0.0337mmol)在二氯甲烷(0.5000ml)中的溶液,并且将该反应混合物保持在0℃搅拌1小时。用etoac(15ml)稀释该反应混合物,并且用0.4m hcl水溶液(10ml)和盐水(15ml)洗涤。分离有机层,经na2so4干燥,过滤,并且真空浓缩。真空浓缩该反应混合物,然后通过硅胶反相快速色谱(biotage系统),使用snap c-18 12g柱纯化,用水:乙腈95:5-40:60洗脱。采集期望的级分,并且真空浓缩,得到3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-c]吡啶-2-酮(19mg),为橙色固体。lc/ms:qc_3_min:rt=1.85m/z 387[m+h]+使用与上述方法类似的方法,用适合的二胺替代4-甲基-n2-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]吡啶-2,3-二胺(中间体75)制备下列化合物。在0℃至室温的温度范围进行反应。通过快速色谱(硅胶柱,使用环己烷/etoac或dcm/甲醇作为洗脱液,或c-18柱,使用水/乙腈作为洗脱液)纯化终产物。实施例393-[2-[(3,3,7-三甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮将3-(2-氯嘧啶-5-基)-1h-咪唑并[4,5-b]吡啶-2-酮(中间体73,40mg,0.1615mmol)、3,3,7-三甲基-2h-苯并呋喃-4-醇(中间体184wo2012076877,80mg,0.2244mmol)和碳酸二钾(碳酸钾)(48mg,0.3473mmol)在二甲亚砜(5ml)中的混合物在120℃搅拌2小时。然后用水(20ml)稀释该混合物,并且用etoac(15ml×3)萃取。用盐水(30ml)洗涤合并的有机层,分离,经mgso4干燥,过滤,并且真空浓缩。然后通过快速柱色谱(biotage),使用modus12g硅胶柱纯化粗残留物,用在dcm中的0-100%etoac洗脱。采集期望的级分,并且浓缩。然后再通过反相柱色谱(biotage),使用snap 30g c-18柱纯化该残留物,用在水中的5-95%乙腈洗脱。采集期望的级分,并且浓缩,得到3-[2-[(3,3,7-三甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮(7mg),为白色固体。lc/ms:qc_3_min:rt=2.01m/z 390[m+h]+生物学实施例生物学实施例1:kv3通道调节的测定本发明化合物调节电压门控钾通道亚型kv3.4/kv3.3/kv3.2/kv3.1的能力可以使用以下测定法来测定。可以使用类似的方法研究本发明化合物调节其它通道亚型的能力。细胞生物学为了评估化合物对人kv3.3通道(hkv3.3)的作用,用pbacmire_kcnc-3载体转染中国仓鼠卵巢(cho)-k1细胞产生表达人kv3.3通道的稳定细胞系。将细胞在补充有10%胎牛血清(gibco)、1×非必需氨基酸(invitrogen)和遗传霉素(g418)400μg/ml的dmem/f12(gibco)中培养。使细胞在37℃在空气中包含5% co2的加湿环境中生长并且维持。为了评估化合物对人kv3.2通道(hkv3.2)的作用,通过用pcih5-hkv3.2载体转染cho-k1细胞来产生表达人kv3.2通道(hkv3.2)的稳定细胞系。在补充有10%胎牛血清、1×非必需氨基酸(invitrogen)和500μg/ml潮霉素-b(invitrogen)的dmem/f12培养基中培养细胞。使细胞在37℃在空气中包含5% co2的加湿环境中生长并且维持。为了评估化合物对人kv3.1通道(hkv3.1)的作用:通过用具有人kv3.1(nm_004976.4)的表达载体转染hek-293细胞来产生人胚肾(hek)-hkv3.1细胞系。用补充有10%热灭活的fbs、2mm l-谷氨酰胺、1%青霉素-链霉素和0.6mg/ml遗传霉素(g418)的mem中培养细胞。使用包含g418选择抗生素(0.6mg/ml)的mem扩增培养基,在t175 cm2培养瓶中在37℃与5%co2下扩增hek-hkv3.1b细胞。每3-4天使细胞脱粘附,使用dpbs洗涤两次培养瓶,然后用tryple使细胞离壁,并且以2-4×106个细胞/瓶的密度重新铺板。为了评估化合物对人kv3.4通道(hkv3.4)的作用:通过用具有人kv3.4(nm_004978)的表达载体转染hek-293细胞来产生人胚肾(hek)-hkv3.4细胞系。用补充有10%热灭活的fbs、2mm l-谷氨酰胺、1%青霉素-链霉素和0.6mg/ml遗传霉素(g418)的mem培养细胞。将hek-hkv3.4细胞在t175 cm2烧瓶中在37℃与5%co2下使用含有g418选择抗生素(0.6mg/ml)的mem扩增培养基扩增。每3-4天使细胞脱粘附,使用dpbs洗涤两次培养瓶,然后用tryple使细胞离壁,并且以4-8×106个细胞/瓶的密度重新铺板。用于ionworks quattrotm试验的细胞制备在本试验的当天,从温育箱中取出细胞并且除去培养基。用5ml不含钙和镁的dulbecco pbs(dpbs)洗涤细胞,并且通过加入3ml versene(invitrogen,italy)脱粘附,然后在37℃短暂温育5分钟。轻敲培养瓶以使细胞离壁,并且加入10ml包含钙和镁的dpbs以制备细胞悬浮液。然后将细胞悬浮液置于15ml离心管中,并且以1200rpm离心2分钟。离心后,除去上清液,并且使用5ml移液管将细胞沉淀重悬于4ml包含钙和镁的dpbs中以分散沉淀。然后校正细胞悬浮液体积以得到用于测定的约300万个细胞/ml的细胞浓度。将添加到细胞中的所有溶液预温热至37℃。电生理学试验在室温进行,使用具有patchplatetm ppc的ionworks quattrotm平面阵列电生理学技术(molecular devices corp.)。使用微型计算机(dell pentium 4)进行刺激方案和数据收集。通过在每个孔上施加10mv电压阶跃来确定平面电极孔电阻(rp)。这些测量在细胞添加之前进行。在细胞添加和密封形成之后,通过施加从-80mv至-70mv的电压阶跃持续160ms来进行密封测试。此后,将两性霉素-b溶液添加到电极的胞内表面以实现胞内进入。将细胞保持在-70mv。在所有试验中,通过施加50ms超极化(10mv)前脉冲以诱导漏电流,然后在测试脉冲之前在维持电位下进行20ms期限来进行漏扣除。对于hkv3.2和hkv3.1,从-70mv的维持电位测定,施加-15mv的第一测试脉冲100ms,并且在-70mv的100ms之后施加+40mv的第二脉冲50ms。然后将细胞在-100mv下维持100ms,并且施加从-70mv至+40mv(持续时间50ms)的另一脉冲以在200ms期间将电压钳位在-40mv。对于hkv3.3测定,从-70mv的维持电位开始,施加第一测试脉冲至0mv,持续500ms,并且在-70mv下再施加100ms,施加第二脉冲至40mv,持续200ms。这些较长的测试脉冲用于研究hkv3.3通道的失活。测试脉冲方案可以在不存在(加入前读数)和存在(加入后读数)测试化合物的情况下进行。可以通过添加化合物、然后温育3分钟来分隔加入前读数和加入后读数。对于hkv3.4,来自-70mv的保持电位的测定,施加-15mv的第一测试脉冲100ms,并且在-70mv的200ms后,施加0mv的第二脉冲100ms,然后在-70mv的200ms后,在200ms期间施加+40mv的第三脉冲。溶液和药物胞内溶液包含如下(以mm计):葡萄糖酸钾100、kcl 54、mgcl2 3.2,hepes 5,用koh调节至ph 7.3。将两性霉素-b溶液制备成在dmso中的50mg/ml储备溶液,并且在胞内溶液中稀释至0.1mg/ml的最终工作浓度。外部溶液是dulbecco磷酸盐缓冲盐水(dpbs)并且包含如下(以mm计):cacl2 0.90、kcl 2.67、kh2po4 1.47、mgcl.6h2o 0.493、nacl 136.9、na3po48.06,ph 7.4。将用于本发明的化合物(或参比化合物例如n-环己基-n-[(7,8-二甲基-2-氧代-1,2-二氢-3-喹啉基)甲基]-n’-苯基脲)以10mm的储备浓度溶于二甲亚砜(dmso)。使用biomek fx(beckman coulter)在384孔化合物板中将这些溶液进一步用dmso稀释。将每个稀释液(1μl)转移到另一个化合物板中,并且加入包含0.05%普流尼克酸(66μl)的外部溶液。加入3.5μl来自每个板的包含本发明化合物,并且在ionworks quattrotm试验期间与细胞一起温育。最终测定稀释度为200,并且最终化合物浓度范围为50μm-50nm。数据分析在化合物不存在下,使用密封电阻(>20mω)和峰值电流振幅(>500pa,在40mv的电压阶跃下)分析和过滤记录,以从进一步分析中消除不适合的细胞。对于hkv3.2和hkv3.1测定,使用对-15mv电压阶跃测量的药物添加前后之间的诱导电流的配对比较来确定每种化合物的正调节作用。测量kv3通道介导的外向电流,由在-15mv电压脉冲的最后10ms内的电流的平均振幅减去在-15mv阶跃之前的10ms期限内-70mv的平均基线电流来确定。然后将加入测试化合物后的这些kv3通道电流与加入化合物前记录的电流进行比较。将数据对参比化合物(50μm n-环己基-n-[(7,8-二甲基-2-氧代-1,2-二氢-3-喹啉基)甲基]-n’-苯基脲)的最大作用和溶媒对照(0.5%dmso)的作用进行归一化。使用activitybase或excel软件分析归一化的数据。通过使用activitybase中的四参数逻辑函数拟合浓度-响应数据来确定将电流由参比化合物产生的最大增加而增加50%所需的化合物浓度(ec50)。对于hkv3.3测定,考虑到在0mv测试脉冲(500ms)的持续时间内峰值电流和电流衰减(失活),针对0mv阶跃测量药物添加前后之间的诱导电流的配对比较。n-环己基-n-[(7,8-二甲基-2-氧代-1,2-二氢-3-喹啉基)甲基]-n’-苯基脲得自asinex(登记号:552311-06-5)。结果表1中提供了许多参比实施例的结构。参比实施例1具有与中心吡啶基环连接的乙内酰脲,而参比实施例2-8具有与中心吡啶基环连接的多种替代杂环基团。在上述测定中测试的所有参比实施例1-8的数据也显示在表1中。参比实施例1如wo2012/076877中概述的合成。使用与本文公开的那些类似的方法合成参比实施例2-9。表1:kv3.1测定结果表1中的数据表明,将乙基取代的乙内酰脲环(参比实施例1)修饰为相应的乙基取代的脲(参比实施例2)或含稠合氧的5,5-双环脲(参比实施例3)导致kv3.1测定中的pec50显著降低和max r特性。类似地,将参比实施例4的乙内酰脲环修饰为相应的二氢尿嘧啶(参比实施例5)和偕二甲基异构体(参比实施例6)导致kv3.1测定中的pec50显著降低和max r特性。去除脲或二氢尿嘧啶的取代导致效力进一步降低(参比实施例7和8)。在上述测定中测试的所有实施例化合物的数据如表2中所示。表2:kv3.1测定结果*数据四舍五入到小数点一位表2中的数据表明,与5-或6-元杂芳环稠合的5-元脲在kv3.1测定中产生显示出良好pec50特性的化合物。包含与3-吡啶基环稠合的5-元脲或与6-元杂环稠合的6-元脲的参比实施例及其相应的实施例化合物的数据如表3中所示。表3:kv3.1测定结果*数据四舍五入到小数点一位表3中的数据表明,包含与3-吡啶基稠合的5-元脲的参比实施例9的效力低于其相应的1-吡啶基化合物(实施例33)和2-吡啶基化合物(实施例23)。包含与3-吡啶基稠合的5-元脲的参比实施例10的效力低于其相应的1-吡啶基化合物(实施例7)。包含与1-吡啶基稠合的6-元脲的参比实施例11的效力低于其相应的与1-吡啶基稠合的5-元脲(实施例1)和与4-吡啶基稠合的5-元脲(实施例36)。生物学实施例1中所述的hkv3.1、hkv3.2和hkv3.3测定的数据的二次分析可用于研究化合物对从去极化电压脉冲开始的电流上升速率的影响。化合物的作用等级可以根据时间常数(tauact)确定,(tauact)使用下面给出的等式从在-15mv去极化电压脉冲开始之后的kv3.1、kv3.2和kv3.3电流的上升的非线性拟合获得。y=(y0-ymax)*exp(-k*x)+ymax其中:y0为去极化电压脉冲开始时的电流值;ymax为坪值电流;k为速率常数,并且tauact为激活时间常数,其为k的倒数。类似地,也可以研究化合物对在-15mv去极化电压脉冲结束时通道闭合时kv3.1、kv3.2或kv3.3电流衰减所花费的时间的影响。在后一种情况下,化合物对通道关闭的影响的等级可以根据在去极化电压脉冲结束后即刻的电流(“尾电流”)衰减的非线性拟合的时间常数(taudeact)确定。kv3.1、kv3.2和kv3.3通道必须非常快速地激活和失活,以便使神经元以高频率激发动作电位(rudy等人,2001)。激活的减慢可能延迟动作电位复极化的启动;失活的减慢可能产生超极化电流,其降低神经元的兴奋性并且延迟神经元可以激发进一步的动作电位之前的时间。这两种对通道激活和失活的减慢作用一起可能导致神经元高频激发的能力降低而不是促进。因此,对kv3.1和/或kv3.2和/或kv3.3通道具有这种减慢作用的化合物将有效地表现为通道的负调节剂,导致神经元激发减慢。对于wo2011/069951中公开的某些化合物,已经显示了后一种作用,其中tauact的显著增加可以从使用电生理学技术在体外大鼠脑皮质中的“快速激发”的中间神经元进行的记录中观察到。加入相关化合物降低了神经元响应于300hz的去极化脉冲串而激发的能力。因此,尽管某些化合物可以在生物学实施例1的重组细胞测定中被鉴定为正调节剂,但是显著增加tauact值的那些化合物可以降低天然组织中神经元高频激发的能力。生物学实施例2:体内药代动力学参数的测定材料和方法对成年雄性大鼠(charles river,italy)口服给予1mg/kg(5ml/kg,在5%v/vdmso,0.5%w/v hpmc,在水中)和静脉内给予0.5mg/kg(2ml/kg,在5%v/v dmso,40%w/vpeg 400,在盐水中)的试验化合物。口服施用后,在深度异氟烷麻醉下从每只大鼠的门静脉和心脏收集血液样品(每个时间点1只大鼠)。静脉内施用后,从每只大鼠的侧尾静脉收集连续血液样品。在这些动物的剂量施用后0.5小时的单个时间点收集血液和脑样品。在所有情况下,将血液样品收集到edta钾管中。另一组大鼠(每种测试化合物n=1)在如上口服施用1mg/kg测试化合物之前不久接受pgp转运抑制剂埃克瑞达(3mg/kg)的单次静脉内施用。可以使用基于用乙腈沉淀蛋白质,然后用优化的分析方法进行hplc/ms-ms分析的方法测定血液和脑样品的测试化合物浓度。分析使用winnonlin professional 4.1版,使用非房室药代动力学模型分析口服或静脉内给药后不同时间点的血液(表示为ng/ml)和脑(表示为ng/g)中测试化合物的浓度。得到以下参数:静脉内给药:随时间的最大浓度(cmax)、随时间的积分浓度(auc)、清除率(clb)、分布体积(vss)、半衰期(t1/2)和脑/心脏血液浓度@0.5hr。口服给药:cmax、最大浓度时间(tmax)、auc、生物利用度(f%)、吸收分数(fa%)、血脑比(auc b/b)和在埃克瑞达存在下auc b/b的倍数变化。表4:测试化合物auc和浓度 实施例 auc b/b(脑/血液)比 脑/心脏血液浓度@0.5hr 8 <0.1 <0.1 13 0.12 0.13 17 0.45 0.76 24 0.29 0.33 26 <0.1 <0.1 33 0.65 0.63 生物学实施例3:体内药代动力学参数的进一步测定血液和脑组织结合的测定将在使用k3-edta作为抗凝血剂的试验周中采集的sprague dawley大鼠全血用等渗磷酸盐缓冲液1:1(v/v)稀释。将在-20℃冷冻储存的sprague dawley大鼠全脑解冻并且在1:2(w/v)的人工脑脊液(csf)中均化。将适量的测试化合物溶于dmso中,得到10毫摩尔溶液。然后使用在milliq水中的50%乙腈制备进一步的稀释液,以获得166.7微摩尔工作溶液。该工作溶液用于掺入血液以在全血中获得0.5微摩尔的终浓度。类似地,该工作溶液用于掺入脑样品以在全脑中获得5微摩尔的终浓度。从这些掺加的血液和脑制品中,立即提取对照样品(n=3),并且用于计算测试品的初始收率。将150μl不含化合物的缓冲液(用于血液的等渗磷酸盐缓冲液或用于脑的人造csf缓冲液)分配到一半孔中,并且将150μl掺加的基质(血液或脑)加载到另一半孔中,其中两个半孔通过半透膜分隔开。在37℃平衡5小时后,将50μl透析的基质(血液或脑)加入50μl相应的不含化合物的缓冲液中,并且对于缓冲液反之亦然,使得缓冲液与基质(血液或脑)的体积保持相同。然后用300μl含有咯利普兰(正电离模式的对照)或双氯芬酸(负电离模式的对照)作为内标的乙腈通过蛋白质沉淀提取样品,并且以3000rpm离心10分钟。收集上清液(100μl),用在milliq水中的27% acn(200μl)稀释,然后注入hplc-ms/ms或uplc-ms/ms系统中以测定存在的测试化合物的浓度。然后使用以下公式确定血液和脑组织结合:afu=缓冲液/血液或afu=csf/脑其中afu=未结合的表观分数;缓冲液=在缓冲液隔室中测定的分析物/内标比;血液=在血液隔室中测定的分析物/内标比;脑=在脑隔室中测定的分析物/内标比。其中:fucr=校正的未结合分数;d=基质稀释因子(血液d=2,并且脑d=3)。然后:%结合=(1-fucr)×100%未结合=100-%结合体外肝细胞中的代谢稳定性研究本研究的目的在于确定试验项目在大鼠和人肝微粒体中的代谢稳定性。维拉帕米和右美沙芬用作微粒体温育的阳性对照。通过将11.2ml磷酸二氢钾和38.8ml磷酸氢二钾合并,用水稀释至1l来制备温育培养基。将冷冻保存的微粒体解冻并且保持在冰上直至使用。然后在温育中将微粒体稀释至0.56mg/ml的蛋白质浓度。离心细胞,重悬于培养基中,并且通过血细胞计数器计数。使用台盼蓝排除测试测量细胞活力。通过将1.7mg的nadp、7.8mg的g6p和6个单位的g6p-脱氢酶溶于1ml的2%碳酸氢钠溶液(通过将20g nahco3溶于1l水来制备)来制备nadph再生系统溶液。将测试化合物分别溶于dmf中以获得10mm储备溶液,将其在水/乙腈50/50(v/v)中进一步稀释以获得相应的50μm工作溶液。将维拉帕米和右美沙芬溶于dmf以获得10mm维拉帕米溶液和10mm右美沙芬溶液。然后用温育培养基稀释这些溶液,以便得到50μm维拉帕米工作溶液和50μm右美沙芬工作溶液。将800μl等份的nadph再生系统在37℃预温热5分钟。将5μl的50μm测试化合物、维拉帕米或右美沙芬加入到445μl的0.56mg/ml微粒体溶液中,并且将温育混合物在96深孔2ml板(温育板)中于37℃预温热5分钟。通过向温育混合物中加入50μl预温热的nadph再生系统启动温育反应。在0、3、6、9、15和30分钟从温育混合物中取出50μl等分试样,并且通过加入100μl具有相应的内标的乙腈终止反应。然后将样品用120μl水(最终有机溶剂百分比为37%)稀释,并且在lc-ms/ms分析之前以3000rpm离心10分钟。由剩余测试化合物与内标物的峰面积比相对于时间计算代谢稳定性。使用温育的实际体积v(ml)、温育中肝细胞的量m(百万个细胞)和每g肝脏的肝细胞数量hn(对于人为120),按照一级消除常数k(min-1)(通过绘制剩余测试品与内标的峰面积比的自然对数相对于时间的曲线,从graphpad获得)确定固有清除率(clint)。clint的值表示为ml/分钟/g肝脏,如表5中所示:表5:测试化合物药代动力学参数*基于wo2021214090中报道的ec50值为5.9。通过与实施例类似的方法制备参比实施例12-lc/ms:qc_3_min:rt=2.04分钟m/z 404[m+h]+。**基于wo2021214090中报道的ec50值为2.4。通过与实施例类似的方法制备参比实施例13-lc/ms:qc_3_min:rt=2.07分钟m/z 403[m+h]+。表5中的数据显示,实施例17显示出多种期望的性质-同时维持良好的效力(pec506.0),实施例17还具有良好的未结合分数(在血液和脑中为1.6%)和良好的清除率(在人和大鼠中cli<0.5)。这些特性与化合物例如实施例4和32以及参比实施例12和13(wo2021214090的实施例25和27)相比是有利的。制剂实施例1-片剂:式(i)化合物或其药学上可接受的盐和/或溶剂化物和/或衍生物可以以本身已知的方式用作活性成分,用于制备具有以下组成的片剂:制剂实施例2-胶囊剂:式(i)化合物或其药学上可接受的盐和/或溶剂化物和/或衍生物可以以本身已知的方式用作活性成分,用于制备具有以下组成的胶囊剂:另外的动物模型专利申请wo2011/069951、wo2012/076877、wo2012/168710、wo2013/175215、wo2013/182851、wo2013/083994、wo2013/182850、wo2017/103604、wo2018/020263、wo2018/109484和wo2020/079422(全部通过引用并入)证明了作为kv3.1和kv3.2的调节剂的化合物在癫痫发作、多动、睡眠障碍、精神病、听力障碍、双相情感障碍和疼痛的动物模型中的活性。专利申请wo2013/175211(通过引用并入)证明了作为kv3.1和kv3.2的调节剂的化合物在栗鼠类急性噪声诱导的听力损失模型中的功效,并且还评估了化合物在中枢听觉处理缺陷模型和耳鸣模型中的功效。在整个说明书和随后的权利要求书中,除非上下文另有要求,否则词语“包含”以及例如“含有”和“包括”的变化形式将被理解为暗示包括所述整数、步骤、整数组或步骤组,但不排除任何其它整数、步骤、整数组或步骤组。本技术包括说明书和权利要求书,其可以用作关于任何后续申请的优先权的基础。这样的后续申请的权利要求可以涉及本文描述的任何特征或特征的组合。它们可以采取产品、组合物、方法或用途权利要求的形式,并且可以包括例如但不限于以下权利要求。本发明的方案方案1.式(i)化合物:其中:v为基团(va)、基团(vb)或基团(vc);其中基团(va)和基团(vb)为:其中:r1为h、c1-4烷基、卤素、卤代c1-4烷基、cn、c1-4烷氧基或卤代c1-4烷氧基;r2为h、c1-4烷基、c3-5螺碳环基、卤代c1-4烷基或卤素;r3为h、c1-4烷基、卤代c1-4烷基、卤素;或r3不存在;r13为h、c1-4烷基、卤代c1-4烷基、卤素;或r13不存在;r14为h、c1-4烷基、卤代c1-4烷基、卤素;或r14不存在;a为具有至少一个o原子的5或6元饱和或不饱和杂环;当与苯基一起考虑时,所述杂环任选与环丙基或环丁基或环戊基稠合以形成三环;其中r2和r3可以连接至相同或不同的环原子;r2可以连接至稠合的环原子;并且其中r13和r14可以连接至相同或不同的环原子;其中基团(vc)为:其中:r16为卤素、c1-4烷基、c1-4烷氧基、卤代c1-4烷基、卤代c1-4烷氧基或cn;r17为h、卤素、cn、c1-4烷基、c1-4烷氧基或卤代c1-4烷氧基;r18为h、卤素、cn、c1-4烷基或c1-4烷氧基;w为n或ch;x为n或ch;y为n或ch;其中w、x和y的至少一个为ch,并且当x和y之一为n时,另一个为ch;z为包含一个或两个氮原子的5-元杂芳基,并且其中氮原子之一和碳原子之一可以独立地任选被甲基取代;或z为包含一个或两个氮原子的6-元杂芳基,其中碳原子之一可以任选被甲基取代;并且条件是z不为或其盐和/或溶剂化物和/或衍生物。方案2.式(i)化合物:其中:v为基团(va)、基团(vb)或基团(vc);其中基团(va)和基团(vb)为:其中:r1为h、c1-4烷基、卤素、卤代c1-4烷基、cn、c1-4烷氧基或卤代c1-4烷氧基;r2为h、c1-4烷基、c3-5螺碳环基、卤代c1-4烷基或卤素;r3为h、c1-4烷基、卤代c1-4烷基、卤素;或r3不存在;r13为h、c1-4烷基、卤代c1-4烷基、卤素;或r13不存在;r14为h、c1-4烷基、卤代c1-4烷基、卤素;或r14不存在;a为具有至少一个o原子的5或6元饱和或不饱和杂环;当与苯基一起考虑时,所述杂环任选与环丙基或环丁基或环戊基稠合以形成三环;其中r2和r3可以连接至相同或不同的环原子;r2可以连接至稠合的环原子;并且其中r13和r14可以连接至相同或不同的环原子;其中基团(vc)为:其中:r16为卤素、c1-4烷基、c1-4烷氧基、卤代c1-4烷基、卤代c1-4烷氧基或cn;r17为h、卤素、cn、c1-4烷基、c1-4烷氧基或卤代c1-4烷氧基;r18为h、卤素、cn、c1-4烷基或c1-4烷氧基;w为n或ch;x为n或ch;y为n或ch;其中w、x和y的至少一个为ch,并且当x和y之一为n时,另一个为ch;z为包含一个或两个氮原子的5-元杂芳基,并且其中氮原子之一和碳原子之一可以独立地任选被甲基取代;或z为包含一个或两个氮原子的6-元杂芳基,其中碳原子之一可以任选被甲基取代;并且条件是z不为或其药学上可接受的盐和/或溶剂化物和/或衍生物。方案3.方案2的化合物或其药学上可接受的盐和/或溶剂化物。方案4.方案3的化合物或其溶剂化物。方案5.方案2的化合物或其药学上可接受的盐。方案6.方案1或2的化合物。方案7.方案1-6任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为基团(va)。方案8.方案1-6任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为基团(vb)。方案9.方案8的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(vb)为:方案10.方案1-9任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r1为h、c1-4烷基、卤素、卤代c1-4烷基或cn,特别是c1-4烷基,例如甲基。方案11.方案10的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r1为h。方案12.方案10的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r1为甲基。方案13.方案1-12任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中环a选自:其中表示环a与苯环稠合的点。方案14.方案13的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中环a选自:方案15.方案14的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中环a为:方案16.方案1-15任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r2为h、c1-4烷基、c3-5螺碳环基或卤素;特别是c1-4烷基或c3-5螺碳环基。方案17.方案16的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r2为c3螺碳环。方案18.方案17的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r2为甲基或卤素,例如氟。方案19.方案18的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r2为甲基。方案20.方案1-19任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r3为h、c1-4烷基、卤代c1-4烷基或卤素。方案21.方案20的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r3为甲基或卤素,例如氟。方案22.方案21的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r3为甲基。方案23.方案1-22任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r13为h或不存在,并且适合地不存在。方案24.方案1-23任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r14为h或不存在,并且适合地不存在。方案25.方案1-6任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为基团(vc)。方案26.方案1-6或25任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r16为c1-4烷基、c1-4烷氧基、卤代c1-4烷基、卤代c1-4烷氧基或cn。方案27.方案26的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r16为甲基、乙基、丙基、丁基、环丙基、氯、氟、甲氧基、乙氧基、丙氧基、三氟甲基、三氟甲氧基或cn。方案28.方案27的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r16为三氟甲氧基或甲氧基。方案29.方案1-6或25-28任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r16在间位上。方案30.方案1-6或25-29任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r17为h、卤素、cn、c1-4烷基或c1-4烷氧基。方案31.方案30的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r17为h。方案32.方案31的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r17为甲基、乙基、丙基、丁基、环丙基、氯、氟、甲氧基、乙氧基、丙氧基、三氟甲氧基或cn。方案33.方案32的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r17为甲基或cn。方案34.方案1-6或30-33任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r17在对位上。方案35.方案1-6或25-34任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r18为h。方案36.方案1-6或25-29任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r17为h,并且r18为h。方案37.方案36的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中r16在间位上。方案38.方案1-6或25-37任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为:方案39.方案1-6或25-37任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为:方案40.方案1-6或25-37任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为:方案41.方案1-6或25-37任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为:方案42.方案1-24任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为:方案43.方案1-24任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为:方案44.方案1-24任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为:方案45.方案1-24任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中v为:方案46.方案1-45任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中w为n,并且x和y为ch。方案47.方案1-45任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中w为n,x为n,并且y为ch。方案48.方案1-45任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中w为n,x为ch,并且y为n。方案49.方案1-45任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中w和x为ch,并且y为n。方案50.方案1-49任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中z为基团(za):方案51.方案50的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中b1为n。方案52.方案50的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中b2为n.方案53.方案50的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中b3为n。方案54.方案50的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中b4为n。方案55.方案50的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(za)选自:方案56.方案50的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(za)选自:方案57.方案55的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(za)选自:方案58.方案56的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(za)选自:方案59.方案57的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(za)选自:方案60.方案58的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(za)选自:方案61.方案59的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(za)为:方案62.方案60的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(za)为:方案63.方案1-49任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中z为基团(zb):其中:c1和c3各自独立地选自ch、c(me)和n;并且c2为nh或n(me);并且其中当c1和c3之一为n时,另一个为ch或c(me)。方案64.方案63的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(zb)选自:方案65.方案63的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(zb)选自:方案66.方案63的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(zb)选自:方案67.方案66的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(zb)选自:方案68.方案67的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(zb)为:方案69.方案68的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(zb)为:方案70.方案1-49任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中z为基团(zc):其中:c2为n、ch或c(me),并且c3为ch或c(me);其中当c2或c3之一为c(me)时,另一个为ch。方案71.方案70的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中zc选自:方案72.方案1-49任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中z为基团(zd):其中:c1为ch或c(me),并且c2为n、ch或c(me);其中当c1或c2之一为c(me)时,另一个为ch。方案73.方案72的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(zd)选自:方案74.方案1-49任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中z为基团(ze-a):其中:e1为ch或cme。方案75.方案74的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(ze-a)选自:方案76.方案1-49任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中z为基团(ze-b):其中e1为ch或c(me)。方案77.方案76的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(ze-b)选自:方案78.方案1-49任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中z为基团(ze-c):其中:e1为ch或c(me)。方案79.方案76的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其中(ze-c)选自:方案80.方案1-6任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其选自:3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;7-甲基-3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-[6-[4-甲基-3-(三氟甲氧基)苯氧基]-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;7-甲基-3-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-2-吡啶基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-[5-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]吡嗪-2-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[5-[3-(三氟甲氧基)苯氧基]吡嗪-2-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[5-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基吡嗪-2-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[6-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-3-吡啶基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-(2-{2h-螺[1-苯并呋喃-3,1’-环丙烷]氧基}嘧啶-5-基)-1h,2h,3h-咪唑并[4,5-b]吡啶-2-酮;4-[[5-(2-氧代-1h-咪唑并[4,5-b]吡啶-3-基)-2-吡啶基]氧基]-2-(三氟甲氧基)苄腈;7-甲基-3-(2-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基嘧啶-5-基)-1h-咪唑并[4,5-b]吡啶-2-酮;3-[2-(3-甲氧基苯氧基)嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[2-[3-(三氟甲氧基)苯氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;2-甲基-6-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-4h-咪唑并[4,5-c]吡唑-5-酮;2-甲基-6-[6-(7-甲基螺[2h苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-4h-咪唑并[4,5-c]吡唑-5-酮;6-[6-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-3-吡啶基]-2-甲基-4h-咪唑并[4,5-c]吡唑-5-酮;2-甲基-6-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-基)-4h-咪唑并[4,5-c]吡唑-5-酮;2-甲基-6-(2-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基嘧啶-5-基)-4h-咪唑并[4,5-c]吡唑-5-酮;3-[2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-c]吡啶-2-酮;2-甲基-9-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]-7h-嘌呤-8-酮;2-甲基-9-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-7h-嘌呤-8-酮;2-甲基-9-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-7h-嘌呤-8-酮;2-甲基-9-[6-[4-甲基-3-(三氟甲氧基)苯氧基]-3-吡啶基]-7h-嘌呤-8-酮;9-[6-(3-甲氧基苯氧基)-3-吡啶基]-2-甲基-7h-嘌呤-8-酮;9-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-7h-嘌呤-8-酮;9-[6-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]-3-吡啶基]-2-甲基-7h-嘌呤-8-酮;3-[2-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[2-[4-甲基-3-(三氟甲氧基)苯氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;3-[2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮;6-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-2,4-二氢咪唑并[4,5-c]吡唑-5-酮;3-[6-(7-甲基螺[2h-苯并呋喃-3,1’-环丙烷]-4-基)氧基-3-吡啶基]-1h-咪唑并[4,5-c]吡啶-2-酮;1-(6-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基-3-吡啶基)-3h-咪唑并[4,5-b]吡啶-2-酮;5-甲基-3-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-基)-1h-咪唑并[4,5-b]吡啶-2-酮;6-甲基-3-(5-螺[2h-苯并呋喃-3,1’-环丙烷]-4-基氧基吡嗪-2-基)-1h-咪唑并[4,5-b]吡啶-2-酮;和3-[2-[(3,3,7-三甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮。方案81.方案1-6任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其为3-(2-{2h-螺[1-苯并呋喃-3,1’-环丙烷]氧基}嘧啶-5-基)-1h,2h,3h-咪唑并[4,5-b]吡啶-2-酮:方案82.方案1-6任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其为3-[2-[3-(三氟甲氧基)苯氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮:方案83.方案1-6任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其为2-甲基-9-[6-[3-(三氟甲氧基)苯氧基]-3-吡啶基]-7h-嘌呤-8-酮:方案84.方案1-6任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物,其为3-[2-[(3,3-二甲基-2h-苯并呋喃-4-基)氧基]嘧啶-5-基]-1h-咪唑并[4,5-b]吡啶-2-酮:方案85.方案81-84任一项的化合物,其中该化合物为药学上可接受的盐形式。方案86.方案81-84任一项的化合物,其中该化合物不为盐的形式。方案87.药物组合物,其包含方案1-86任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物以及药学上可接受的载体或赋形剂。方案88.方案1-86任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物或方案87的药物组合物,其用作药物。方案89.方案1-86任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物或方案87的用于所述用途的药物组合物,其用于预防或治疗选自以下的疾病或障碍:听力障碍、精神分裂症、抑郁和心境障碍、双相情感障碍、物质滥用障碍、焦虑障碍、睡眠障碍、听觉过敏和响度感知障碍、梅尼埃病、平衡障碍和内耳障碍、冲动控制障碍、人格障碍、注意力缺陷/多动障碍、孤独症谱系障碍、进食障碍、认知损伤、共济失调,疼痛例如神经性疼痛、炎性疼痛和混杂的疼痛,路易体痴呆和帕金森病。方案90.方案1-86任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物或用于方案87的用于所述用途的药物组合物,其用于预防或治疗进行性肌阵挛性癫痫(包括与kcnc1基因突变相关的pme)、听力障碍(包括听力损失和耳鸣)、脆性x染色体综合征、精神分裂症、物质滥用障碍或疼痛。方案91.方案1-86任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物或方案87的用于所述用途的药物组合物在制备药物中的用途。方案92.方案91的用途,其用于制备用于预防或治疗进行性肌阵挛性癫痫(包括与kcnc1基因突变相关的pme)、听力障碍(包括听力损失和耳鸣)、脆性x染色体综合征、精神分裂症、物质滥用障碍或疼痛的药物。方案93.用于预防或治疗进行性肌阵挛性癫痫(包括与kcnc1基因突变相关的pme)、听力障碍(包括听力损失和耳鸣)、脆性x染色体综合征、精神分裂症、物质滥用障碍或疼痛的方法,该方法包括向个体施用方案1-86任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物或方案87的用于所述用途的药物组合物。方案94.方案1-93任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防或治疗进行性肌阵挛性癫痫。方案95.方案1-91任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防或治疗与kcnc1基因突变相关的进行性肌阵挛性癫痫。方案96.方案94的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防或治疗青少年进行性肌阵挛性癫痫。方案97.方案1-93任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防或治疗听力损失。方案98.方案1-93任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防或治疗耳鸣。方案99.方案1-93任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防或治疗脆性x染色体综合征。方案100.方案1-93任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防或治疗精神分裂症。方案101.方案1-93任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防或治疗物质滥用障碍。方案102.方案1-93任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防或治疗疼痛,例如神经性疼痛、炎性疼痛或混杂的疼痛。方案103.方案1-102任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于预防。方案104.方案1-103任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于治疗。方案105.方案1-104任一项的衍生物,在脲的仲氮上被基团l官能化,其中l选自:-po(oh)o-·m+,其中m+为药学上可接受的一价抗衡离子,-po(o-)2·2m+,-po(o-)2·d2+,其中d2+为药学上可接受的二价抗衡离子,-ch(rx)-po(oh)o-·m+,其中rx为氢或c1-3烷基,-ch(rx)-po(o-)2·2m+,-ch(rx)-po(o-)2·d2+,和-co-ch2ch2-co2·m+。方案106.方案1-105任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于人。方案107.方案106的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于小于18岁的人。方案108.方案107的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于4至17岁的人。方案109.方案106的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于18至65岁的人。方案110.方案106的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于66岁或以上年龄的人。方案111.方案1-110任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于以5mg/天至250mg/天施用。方案112.方案1-111任一项的化合物、其药学上可接受的盐、其溶剂化物和/或其衍生物、药物组合物、用途或方法,其用于施用至少三个月的时间期限。方案113.化合物或其盐,选自:-式(ii)化合物:其中v、w、x、y和z、b1、b2、b3和b4如对式(i)化合物所定义;-式(iii)化合物:其中v、w、x、y和z如对式(i)化合物所定义,并且d为卤素,例如cl、br或i;-式(iv)化合物:其中w、x、y和z如对式(i)化合物所定义,并且e为卤素,例如f或cl;-式(vi)化合物:其中v、w、x、y和z如对式(i)化合物所定义;-式(ix)化合物:其中w、x、y和z如对式(i)化合物所定义,并且e为卤素,例如f或cl;-式(x)化合物:其中w、x、y和z如对式(i)化合物所定义,e为卤素,例如f或cl,并且d为卤素,例如cl、br或i;和-式(xiii)化合物:其中w、x、y和z如对式(i)化合物所定义,并且e为卤素,例如f或cl;或其盐,例如其药学上可接受的盐。方案114.用于制备式(i)化合物或其药学上可接受的盐和/或溶剂化物和/或衍生物的方法,该方法包括使式(ii)化合物:或其盐,其中v、w、x、y和z如对式(i)化合物所定义,与羰基化剂例如三光气或羰基二咪唑反应。方案115.制备式(i)化合物或其药学上可接受的盐和/或溶剂化物和/或衍生物的方法,该方法包括使式(iii)化合物:或其盐,其中v、w、x、y和z如对式(i)化合物所定义,并且d为卤素,例如cl、br或i,在金属催化的交叉偶联条件下反应。方案116.制备式(i)化合物或其药学上可接受的盐和/或溶剂化物和/或衍生物的方法,该方法包括使式(iii)化合物:或其盐,其中w、x、y和z如对式(i)化合物所定义,并且e为卤素,例如f或cl,与式(v)化合物:或其盐反应,其中v如权利要求1中所定义。方案117.制备式(ii)化合物或其盐的方法,该方法包括使式(vi)化合物:或其盐,其中v、w、x、y和z如对式(i)化合物所定义,在还原条件下(例如在f粉和氯化铵存在下)反应。方案118.制备式(iii)化合物或其盐的方法,该方法包括使式(vii)化合物:或其盐,其中v、w、x和y如对式(i)化合物所定义,与式(viii)化合物:或其盐反应,其中z如对式(i)化合物所定义,并且d为卤素,例如cl、br或i。方案119.制备式(iv)化合物或其盐的方法,该方法包括使式(ix)化合物:或其盐,其中w、x、y和z如对式(i)化合物所定义,并且e为卤素,例如cl或f,与羰基化剂,例如三光气或羰基二咪唑反应。方案120.制备式(iv)化合物或其盐的方法,该方法包括使式(x)化合物:或其盐,其中w、x、y和z如对式(i)化合物所定义,e为卤素,例如f或cl,并且d为卤素,例如cl、br或i,在金属催化的交叉偶联条件下反应。方案121.制备式(vi)化合物或其盐的方法,该方法包括使式(vii)化合物:或其盐,其中v、w、x和y如对式(i)化合物所定义,与式(xi)化合物:或其盐反应,其中z如对式(i)化合物所定义,并且d为卤素,例如cl、br或i。方案122.制备式(ix)化合物或其盐的方法,该方法包括使式(xiii)化合物:或其盐,其中w、x、y和z如对式(i)化合物所定义,并且e为卤素,例如f或cl,在还原条件下(例如在fe粉和氯化铵存在下)反应。方案123.制备式(x)化合物或其盐的方法,该方法包括使式(xiv)化合物:或其盐,其中w、x和y如对式(i)化合物所定义,并且e为卤素,例如f或cl,与式(viii)化合物:或其盐反应,其中z如方案1中所定义,并且d为卤素,例如cl、br或i。方案124.制备式(ix)化合物或其盐的方法,该方法包括使式(xiv)化合物:或其盐,其中w、x和y如对式(i)化合物所定义,并且e为卤素,例如f或cl,与式(xi)化合物:或其盐反应,其中z如对式(i)化合物所定义,并且d为卤素,例如cl、br或i。参考文献本说明书中引用的所有出版物,包括但不限于专利和专利申请,通过引用并入本文,如同每个单独的出版物被具体和单独地指出通过引用并入本文,如同完全列出一样。anderson la et al.increased spontaneous firing rates in auditorymidbrain following noise exposure are specifically abolished by a kv3 channelmodulator.hear res.2018 aug;365:77-89 andrade-talavera et al.,j.physiol.(2020)598,3711-3725aroniadou-anderjaska v et al.mechanisms regulating gabaergicinhibitory transmission in the basolateral amygdala:implications for epilepsyand anxiety disorders.amino acids 2007 aug;32:305-315.baranauskas g,nistri a.sensitization of pain pathways in the spinalcord:cellular mechanisms.prog.neurobiol.1998 feb;54(3):349-65.baron r et al.peripheral input and its importance for centralsensitization.ann.neurol.2013 nov;74(5):630-6.ben-ari y.seizure beget seizure:the quest for gaba as a keyplayer.crit.rev.neurobiol.2006;18(1-2):135-144.benes fm et al.circuitry-based gene expression profiles in gaba cellsof the trisynaptic pathway in schizophrenics versus bipolars.pnas 2008 dec;105(52):20935-20940.bennett dl,woods cg.painful and painless channelopathies.lancetneurol,2014 jun;13(6):587-99.berge s et al.pharmaceutical salts.j.pharm.sci.1977;66;1-19.boddum et al.,neuropharm.(2017)118,102-112brambilla p et al.gabaergic dysfunction in mooddisorders.mol.psych.2003 apr;8:721-737.brooke re et al.spinal cord interneurones labelled transneuronallyfrom the adrenal gland by a gfp-herpes virus construct contain the potassiumchannel subunit kv3.1b.auton.neurosci.2002 jun;98(1-2):45-50.brooke re et al.association of potassium channel kv3.4 subunits withpre-and post-synaptic structures in brainstem and spinal cord.neuroscience2004;126(4):1001-10.brooke re et al.immunohistochemical localisation of the voltage gatedpotassium ion channel subunit kv3.3 in the rat medulla oblongata and thoracicspinal cord.brain res.2006 jan;1070(1):101-15.cervero f.spinal cord hyperexcitability and its role in pain andhyperalgesia.exp.brain res.2009 jun;196(1):129-37.chambers ar et al.pharmacological modulation of kv3.1 mitigatesauditory midbrain temporal processing deficits following auditory nervedamage.sci rep.2017dec 13;7(1):17496chang sy et al.distribution of kv3.3 potassium channel subunits indistinct neuronal populations of mouse brain.j.comp.neuro.2007feb;502:953-972.chien ly et al.reduced expression of a-type potassium channels inprimary sensory neurons induces mechanical hypersensitivity.j.neurosci.2007sep;27(37):9855-65.chow a et al.k+channel expression distinguishes subpopulations ofparvalbumin-and somatostatin-containing neocorticalinterneurons.j.neurosci.1999 nov;19(21):9332-9345.darnell et al.,cell 2001,107,489-499desai r et al.protein kinase c modulates inactivation of kv3.3channels.j.biol.chem.2008;283;22283-22294.deuchars sa et al.properties of interneurones in theintermediolateral cell column of the rat spinal cord:role ofthe potassiumchannel subunit kv3.1.neuroscience 2001;106(2):433-46.devulder j.flupirtine in pain management:pharmacological propertiesand clinical use.cns drugs 2010 oct;24(10):867-81.dib-hajj sd et al.the na(v)1.7sodium channel:from molecule toman.nat.rev.neurosci.2013 jan;14(1):49-62.diochot s et al.sea anemone peptides with a specific blockingactivity against the fast inactivating potassium channelkv3.4.j.biol.chem.1998mar;273(12);6744-6749.engel ak et al.dynamic predictions:oscillations and synchrony in top-down processing.nat.rev.neurosci.2001oct;2(10):704-716.ei-hassar et al..j neurosci.201939,4797-4813espinosa f et al.alcohol hypersensitivity,increased locomotion,andspontaneous myoclonus in mice lacking the potassium channels kv3.1 andkv3.3.j.neurosci.2001sep;21(17):6657-6665.espinosa f et al.ablation of kv3.1 and kv3.3 potassium channelsdisrupts thalamocortical oscillations in vitro and in vivo.j.neurosci.2008may;28(21):5570-5581.figueroa k et al.kcnc3:phenotype,mutations,channel biophysics-a studyof 260 familial ataxia patients.human mutation.2010;31;191-196.finnerup nb et al.pharmacotherapy for neuropathic pain in adults:asystematic review and meta-analysis.lancet neurol.2015 feb;14(2):162-73.fisahn a.kainate receptors and rhythmic activity in neuronalnetworks:hippocampal gamma oscillations as a tool.j.physiol.2005 oct;561(1):65-72.glait l et al.effects of aut00063,a kv3.1 channel modulator,on noise-induced hyperactivity in the dorsal cochlear nucleus.hearres.2018 apr;361:36-44greene tw,wuts,pg.greene’s protective groups in organic synthesis,2006,4th edition,john wiley&sons,inc.,hoboken,nj,usa.joho rh et al.increasedγ-and decreasedδ-oscillations in a mousedeficient for a potassium channel expressed in fast-spiking interneurons.j.neurophysiol.1999 jun;82:1855-1864.joho rh,hurlock ec.the role of kv3-type potassium channels incerebellar physiology and behavior.cerebellum 2009feb;8:323-333.jung d et al.age-related changes in the distribution of kv1.1 andkv3.1 in rat cochlear nuclei.neurol,res.2005;27;436-440.kasten mr et al.differential regulation of action potential firing inadult murine thalamocortical neurons by kv3.2,kv1,and sk potassium and n-typecalcium channels.j,physiol.2007;584(2):565-582.kaczmarek l et al.regulation of the timing of mntb neurons by short-term and long-term modulation of potassium channels.hearing res.2005;206;133-145.lau d et al.impaired fast-spiking,suppressed cortical inhibition,andincreased susceptibility to seizures in mice lacking kv3.2 k+channelproteins.j.neurosci.2000 dec;20(24):9071-9085.leger et al.,european neuropsychopharmacology volume 25,supplement 2,september 2015,page s480li w et al.localization of two high-threshol potassium channelsubunits in the rat central auditory system.j.comp.neuro.2001may;437:196-218.lu r et al.slack channels expressed in sensory neurons controlneuropathic pain in mice.j.neurosci.2015jan;35(3):1125-35.markram h et al.interneurons of the neocortical inhibitorysystem.nat.rev.neurosci.2004 oct;5:793-807.martina m et al.functional and molecular differences between voltage-gated k+channels of fast-spiking interneurons and pyramidal neurons of rathippocampus.j.neurosci.1998 oct;18(20):8111-8125.mccarberg bh et al.the impact of pain on quality of life and theunmet needs of pain management:results from pain sufferers and physiciansparticipating in an internet suryey.am.j.ther.2008 jul-aug;15(4):312-20.mcdonald aj,mascagni f.differential expression of kv31b and kv3.2potassium channel subunits in interneurons of the basolateralamygdala.neuroscience 2006;138:537-547.mcmahon a et al.allele-dependent changes of olivocerebellar circuitproperties in the absence of the voltage-gated potassium channels kv3.1 andkv3.3.eur.j.neurosci.2004mar;19:3317-3327.muona m,et al.a recurrent de novo mutation in kcnc1 causesprogressive myoclonus epilepsy.nat genet.2015 jan;47(1):39-46.muqeem t et al.regulation of nociceptive glutamatergic signaling bypresynaptic kv3.4 channels in the rat spinal dorsal horn j neurosci.2018 apr11;38(15):3729-3740olsen t et al.kv3 k+ currents contribute to spike-timing in dorsalcochlear nucleus principal cells.neuropharmacology 2018 may 1;133:319-333parekh et al.,neuropsychopharmacology volume 43,pages 435-444(2018)pilati n et al..acoustic over-exposure triggers burst firing indorsal cochlear nucleus fusitorm cells.hearing research 2012;283;98-106.pirbhoy et al.,journal of neurochemistry 2020 volume 155,issue 5pages 538-558puente n et al.precise localization of the voltage-gated potassiumchannel subunits kv3.1b and kv3.3 revealed in the molecular layer of the ratcerebellar cortex by a pre-embedding immunogold method.histochem.cell.biol.2010sep;134:403-409.reynolds gp et al.calcium binding protein markers of gaba deficits inschizophrenia-post mortem studies and animal models.neurotox.res.2004feb;6(1):57-62.ritter dm et al.modulation of kv3.4 channel n-type inactivation byprotein kinase c shapes the action potential in dorsal root ganglionneurons.j.physiol.2012 jan;590(pt 1):145-61.ritter dm et al.dysregulation of kv3.4 channeis in dorsal rootganglia following spinal cord injury.j.neurosci.2015 jan;35(3):1260-73.roberts l et al.ringing ears:the neuroscience oftinnitus.j.neurosci.2010:30(45);14972-14979.rudy b,mcbain cj.kv3 channels:voltage-gated k+channels designed forhigh-frequency repetitive firing.trends in neurosci.2001 sep;24(9):517-526.sacco t et al.properties and expression of kv3 channels in cerebellarpurkinje cells.mol.cell.neurosci.2006 jul;33:170-179.schulz p,steimer t.neurobiology of circadian systems.cns drugs 2009;23(suppi.2):3-13.song p et al.acoustic environment determines phosphorylationstate of the kv3.1 potassium channel in auditory neuronsnat.neurosci.2005oct;8(10):1335-1342.spencer km et al.neural synchrony indexes disordered perception andcognition in schizophrenia.pnas 2004 dec;101(49):17288-17293.strumbos et al.,j neurosci.201030,10263-10271sun s et al.inhibitors of voltage-gated sodium channel nav1.7:patentapplications since 2010.pharm.pat.anal.2014 sep;3(5):509-21.u.s.department of health and human services,food and drugadministration.draft guidance for industry analgesic indications:developingdrug and biological products:http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/auidances/ucm384691.pdf 2014 feb.von hehn c et al.loss of kv3.1 tonotopicity and alterations in campresponse element-binding protein signaling in cantral auditory neurons ofhearing impaired mice.j.neurosci.2004;24:1936-1940.wickanden ad,mcnaughton-smith g.kv7 channels as targets for thetreatment of pain.curr.pharm.des.2009;15(15):1773-98.woolf cj.what is this thing called pain?j.clin.invest.2010 nov;120(11):3742-4.woolf cj.central sensitization;implications for the diagnosis andtreatment of pain.pain 2011 mar;152(3suppl):s2-15.yanagi m et al.kv3.1-containing k(+)channels are reduced in untreatedschizophrania and normalized with antipsychotic drugs.mol psychiatry.2014.19(5):573-9.yeung sym et al.modulation of kv3 subfamily potassium currents by thesaa anemone toxin bds:significance for cns and biophysicalstudies.j.neurosci.2005 mar;25(38):8735-8745.zamponi gw et al.the physiology,pathology,and pharmacology ofvoltage-gated calcium channels and their future therapeutic potentialpharmacol rev.2015 oct;67(4):821-70。
背景技术:
技术实现思路
- 还没有人留言评论。精彩留言会获得点赞!