WEE1抑制剂及其制备和用途的制作方法
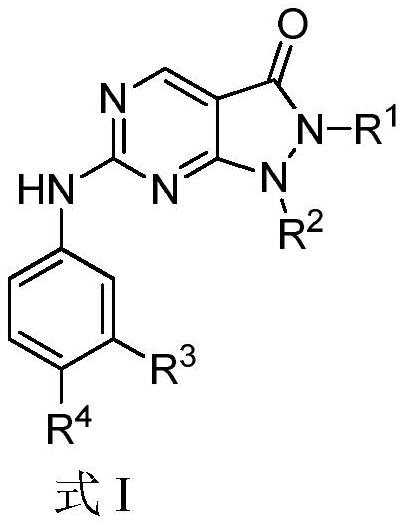
本发明涉及一类具有抑制wee1激酶活性的化合物,以及其在治疗由wee1介导的疾病中的用途。
背景技术:
1、wee1酪氨酸激酶是细胞周期g2期检查点。细胞周期受到严密调控,当细胞的dna未受到损伤时,g1期、s期和g 2期检查点促进细胞进入分裂期,保证细胞周期顺利完成。(clinical cancer research,2011,17(13):4200-4207.)细胞周期受细胞周期蛋白依赖激酶(cdks)调控,cdks家族有14个丝氨酸/苏氨酸蛋白激酶。cdk的活性被磷酸化和结合不同周期蛋白所调控。细胞从g2期到进入分裂期的转换被cdk1(也被称为cdc2)的磷酸化及其相关周期蛋白b正调控。细胞分裂前,cdk1处于非活性状态,它的第15位酪氨酸被wee1磷酸化,然后它的第14位苏氨酸被髓鞘转录因子(myt1)磷酸化。因此,wee1是细胞周期的负调控因子,通过阻止周期蛋白b和激活的cdk1复合物进入细胞核,负调控细胞从g2期进入分裂期。wee1在s期和g2期的表达量和活性都提高,在高度磷酸化的m期降低。当细胞进入g2期,没有dna损伤发生时,polo样蛋白激酶1(plk1)磷酸化wee1,通过泛素连接酶复合物将wee1降解。plk1也磷酸化并且激活蛋白磷酸酶细胞分裂周期25类似物(cdc25),cdc25通过去磷酸化激活cdk1。活性的cdk1可以和周期蛋白b结合,促进细胞进入分裂期。(molecular&cellularbiology,2012,32(20):4226.)
2、当细胞的dna受到损伤时,g1期、s期和g2期检查延迟细胞进入分裂期,为细胞进入分裂之前修复损伤的dna争取了时间,从而保证了基因组的完整性。g1期检查点的关键调控因子p53在很多恶性肿瘤细胞中是突变形式。(proceedings of the national academy ofsciences of the united states of america,2007,104(10):3753-3758.)p53功能缺陷的肿瘤细胞,在dna受到损伤时,不能将细胞周期阻滞在g1期,因此更加依赖于g2期检查点。针对dna损伤,g2期检查点通过平行且互相联系的两条路径抑制cdk1的磷酸化,从而延迟细胞进入分裂期。根据dna损伤类型,共济失调性毛细血管扩张症变异(atm)蛋白激酶或者共济失调性毛细血管扩张相关(atr)蛋白激酶被激活。(oncotarget,2016,7(31):49902-49916.)
3、atm被离子辐射、放射剂和引起双链dna断裂的试剂激活。atm磷酸化和激活检查点激酶2(chk2),chk2磷酸化细胞分裂周期25c磷酸酶(cdc25c)的ser216。这就导致cdc25c的向核输出和细胞质分离,从而抑制它的磷酸化活性。抑制cdc25c的活性导致cdk1/cdk2结合周期蛋白b复合物磷酸化被抑制,使cdk1处于失活形式,抑制细胞进入分裂期。(molecularcancer,2014,13(1):72.)
4、atr由广泛的导致单链dna断裂的基因毒性刺激因素激活的。atr是负责磷酸化和激活chk1的主要激酶。与chk2只能被atm激活相比,chk1可以被atm和atr激活。chk1同时磷酸化wee1和cdc25c,激活wee1激酶活性,抑制cdc25c的磷酸酶活性。wee1磷酸化cdk1-结合周期蛋白b,导致细胞周期阻滞在g2期,为dna修复提供时间。(drug news&perspectives,2010,23(7):425.)
5、wee1在很多恶性肿瘤中过表达,比如肝癌、乳腺癌、恶性胶质瘤、黑色素瘤、成人和儿童脑瘤。其中一部分肿瘤细胞g1检查点异常,如果抑制wee1活性会导致g2期检查点故障,此时带有没有修复的损伤dna的细胞会持续进行分裂,最终分裂致死。(molecular cancertherapeutics,2013,12(12):2675-2684.)无论通过嘧啶衍生物(pd0166285)或者小干扰rna敲低方式,抑制wee1的活性都会使卵巢癌、结肠癌、宫颈癌、骨肉瘤、恶性胶质瘤和肺癌细胞对放射和拓扑异构酶抑制产生的dna损伤更敏感。因此,wee1抑制剂单药和联合用药都具有广阔的发展空间。(cancer biology&therapy,2010,9(7):523-525.)
6、专利wo2007126122,wo2008133866,wo2013012681,wo2013126656,wo2014167347,wo2015092431,wo2018011569,wo2018011570,wo2018090939,wo2018133829,wo2018171633等中描述了具有wee1激酶抑制活性的小分子化合物。目前最领先的化合物是azd1775,已经进入临床ii期试验,显示出良好的癌症治疗效果。
技术实现思路
1、本发明的目的是提供了一种如式i所示的化合物、或其立体异构体、或其药学上可接受的盐:
2、
3、其中,
4、所述的r1选自-c1~6烷基、-c2~6烯基、-c2~6炔基、-c0~2亚烷基-cn、-c0~2亚烷基-(3~10元环烷基)、-c0~2亚烷基-(3~10元杂环烷基);
5、r2选自
6、所述的x选自o、nh或ch2;
7、所述的x1选自ch或n;
8、r21、r22、r29分别独立地选自氢、氘、卤素、氰基、硝基、-oh、-c1~6烷基、卤素取代的c1~6烷基、-c0~2亚烷基-oh、-o(c1~6烷基)、-o(卤素取代的c1~6烷基)、-nh2、-c0~2亚烷基-nh(c1~6烷基)、-c0~2亚烷基-n(c1~6烷基)(c1~6烷基)、-c0~2亚烷基-(3~10元环烷基)、-c0~2亚烷基-(3~10元杂环烷基);
9、所述的r23、r24与其直接相连的原子一起形成3~10元碳环、3~10元杂环;
10、所述的r25、r26与其直接相连的原子一起形成3~10元碳环、3~10元杂环;
11、所述的r27、r28与其直接相连的原子一起形成3~10元碳环、3~10元杂环;
12、r3选自氢、氘、卤素、氰基、硝基、-c1~6烷基、卤素取代的c1~6烷基、-c0~2亚烷基-oh、-o(c1~6烷基)、-o(卤素取代的c1~6烷基)、-nh2、-c0~2亚烷基-nh(c1~6烷基)、-c0~2亚烷基-n(c1~6烷基)(c1~6烷基);
13、所述的r4选自3~12元杂环烷基;所述的杂环烷基可进一步被一个、两个、三个或四个独立的r41取代;
14、所述的r41选自氢、卤素、氰基、硝基、-oh、-c1~6烷基、卤素取代的c1~6烷基、-c0~2亚烷基-oh、-o(c1~6烷基)、-o(卤素取代的c1~6烷基)、-nh2、-c0~2亚烷基-nh(c1~6烷基)、-c0~2亚烷基-n(c1~6烷基)(c1~6烷基)、-c(o)c1~6烷基、3~10元碳环、3~10元杂环;所述碳环、杂环可进一步被一个、两个、三个或四个独立的r31取代;
15、或者,所述的r3、r4与其直接相连的原子一起形成3~10元碳环、3~10元杂环;所述的碳环、杂环烷基可进一步被一个、两个、三个或四个独立的r31取代;
16、所述的r31选自氢、卤素、氰基、硝基、-oh、-c1~6烷基、卤素取代的c1~6烷基、-c0~2亚烷基-oh、-o(c1~6烷基)、-o(卤素取代的c1~6烷基)、-nh2、-c0~2亚烷基-nh(c1~6烷基)、-c0~2亚烷基-n(c1~6烷基)(c1~6烷基)。
17、优选的,本发明所述的化合物、或其立体异构体、或其药学上可接受的盐,所述的r1选自甲基、乙基、
18、优选的,本发明所述的化合物、或其立体异构体、或其药学上可接受的盐,r21、r22、r29分别独立地选自氢、氘、氰基、甲基、乙基、-oh、三氟甲基、环丙基、-ch2oh、-nh2。
19、优选的,本发明所述的化合物、或其立体异构体、或其药学上可接受的盐,所述的r23、r24与其直接相连的原子一起形成环丙基、环丁基、环戊基;
20、所述的r25、r26与其直接相连的原子一起形成环丙基、环丁基、环戊基;
21、所述的r27、r28与其直接相连的原子一起形成环丙基、环丁基、环戊基。
22、优选的,本发明所述的化合物、或其立体异构体、或其药学上可接受的盐,所述的r2选自
23、
24、优选的,本发明所述的化合物、或其立体异构体、或其药学上可接受的盐,所述的r3选自氢、氟、甲基、-ch2oh、甲氧基。
25、优选的,本发明所述的化合物、或其立体异构体、或其药学上可接受的盐,所述的r4选自含氮6元杂环、7元含氮桥环、8元含氮桥环、9元含氮杂螺环、11元含氮杂螺环。
26、进一步地:所述的r4选自
27、进一步地:所述的r4选自
28、优选的,本发明所述的化合物、或其立体异构体、或其药学上可接受的盐,所述的r3、r4与其直接相连的原子一起形成6元含氮杂环。
29、更优选的,本发明所述的化合物、或其立体异构体、或其药学上可接受的盐,所述的r3、r4与其直接相连的原子一起形成
30、进一步地:所述的r31选自甲基。
31、优选的,本发明所述的化合物、或其立体异构体、或其药学上可接受的盐,式i所述的化合物具体为:
32、
33、
34、
35、
36、
37、
38、
39、
40、
41、
42、
43、
44、
45、
46、
47、
48、
49、
50、
51、
52、
53、
54、
55、
56、
57、
58、
59、
60、
61、
62、
63、更优选,本发明的化合物、或其立体异构体、或其药学上可接受的盐,式i所述的化合物具体为:
64、本发明还提供一种上述任一项所述的化合物、或其立体异构体、或其药学上可接受的盐在制备治疗wee1介导的疾病的药物中的用途。
65、所述wee1介导的疾病是与炎症、自身免疫性疾病、感染性疾病、癌症、癌前期综合征相关的疾病中的一种或几种。
66、本发明还提供一种药物组合物,它是以上述任一项所述的化合物、或其立体异构体、或其药学上可接受的盐,加上药学上可接受的辅料制备而成的制剂。
67、以下为对本发明的名词术语的解释和说明:
68、“癌症”或“恶性肿瘤”是指以不受控制的细胞异常增殖为特征的多种疾病中的任何一种,受影响的细胞在局部或通过血流和淋巴系统扩散到其他部位的能力的身体(即转移)以及许多特征结构和/或分子特征中的任何一个。“癌细胞”是指经历多步骤肿瘤进展的早期,中期或晚期阶段的细胞。癌症包括肉瘤、乳腺癌、肺癌、脑癌、骨癌、肝癌、肾癌、结肠癌和前列腺癌。在一些实施方案中,式i的化合物用于治疗选自结肠癌、脑癌、乳腺癌、纤维肉瘤和鳞状细胞癌的癌症。在一些实施方案中,癌症选自黑素瘤、乳腺癌、结肠癌、肺癌和卵巢癌。在一些实施方案中,所治疗的癌症是转移性癌症。
69、自身免疫性疾病是由身体对体内正常存在的物质和组织的免疫反应引起的。自身免疫疾病的例子包括心肌炎、狼疮性肾炎、原发性胆汁性肝硬化、牛皮癣、1型糖尿病、格雷夫氏病、腹腔疾病、克罗恩病、自身免疫性中性白细胞减少症、幼年型关节炎、类风湿性关节炎、纤维肌痛、吉兰巴利综合征、多发性硬化症和自身免疫性视网膜病变。本发明的一些实施方案涉及治疗自身免疫疾病如牛皮癣或多发性硬化症。
70、炎症疾病包括以组织病理性炎症为特征的多种病症。炎性疾病的例子包括寻常性痤疮、哮喘、腹腔疾病、慢性前列腺炎、肾小球性肾炎、炎症性肠病、盆腔炎、再灌注损伤、类风湿性关节炎、结节病、血管炎、房尘螨引起的气道炎症和间质性膀胱炎。炎性疾病与自身免疫性疾病之间存在显著重叠。本发明的一些实施方案涉及炎性疾病哮喘的治疗。免疫系统通常涉及炎症性疾病,在过敏反应和一些肌病中都有表现,许多免疫系统疾病导致异常炎症。il-17a介导的疾病也包括自身免疫性炎症性疾病。
71、本发明中提供的化合物和衍生物可以根据iupac(国际纯粹与应用化学联合会)或cas(化学文摘服务社,columbus,oh)命名系统命名。
72、关于本发明的使用术语的定义:除非另有说明,本文中基团或者术语提供的初始定义适用于整篇说明书的该基团或者术语;对于本文没有具体定义的术语,应该根据公开内容和上下文,给出本领域技术人员能够给予它们的含义。
73、“取代”是指分子中的氢原子被其它不同的原子或分子所替换。
74、碳氢基团中碳原子含量的最小值和最大值通过前缀表示,例如,前缀ca~b烷基表明任何含“a”至“b”个碳原子的烷基。因此,例如,“c1~4烷基”是指包含1~4个碳原子的烷基。
75、“烷基”是指具有指定数目的成员原子的饱和烃链。例如,c1~6烷基是指具有1至6个成员原子,例如1至4个成员原子的烷基基团。烷基基团可以是直链或支链的。代表性的支链烷基基团具有一个、两个或三个支链。烷基基团可任选地被一个或多个如本文所定义的取代基取代。烷基包括甲基、乙基、丙基(正丙基和异丙基)、丁基(正丁基、异丁基和叔丁基)、戊基(正戊基、异戊基和新戊基)和己基。烷基基团也可以是其他基团的一部分,所述其他基团为例如c1~c6烷氧基。
76、“环烷基”、“环烷烃”是指具有碳原子且没有环杂原子且具有单个环或多个环(包括稠合、并和、桥环)的饱和或部分饱和的环状基团。对于具有不含环杂原子的芳族和非芳族环的多环体系,当连接点位于非芳族碳原子时,适用术语“环烷基”(例如5,6,7,8,-四氢化萘-5-基)。术语“环烷基”包括环烯基基团,诸如环己烯基。环烷基基团的实例包括例如,金刚烷基、环丙基、环丁基、环己基、环戊基、环辛基、环戊烯基和环己烯基。包括多双环烷基环体系的环烷基基团的实例是双环己基、双环戊基、双环辛基等。例如
77、“烯基”是指具有2至10个碳原子和在一些实施方案中2至6个碳原子或2至4个碳原子且具有至少1个乙烯基不饱和位点(>c=c<)的直链或支链烃基基团。例如,(ca-cb)烯基是指具有a至b个碳原子的烯基基团并且意在包括例如乙烯基、丙烯基、异丙烯基、1,3-丁二烯基等。
78、“炔基”是指含有至少一个三键的直链一价烃基或支链一价烃基。术语“炔基”还意在包括具有一个三键和一个双键的那些烃基基团。例如,(c2-c6)炔基意在包括乙炔基、丙炔基等。
79、“卤素”为氟、氯、溴或碘。
80、“卤素烷基”指烷基中的氢原子可被一个或多个卤素原子取代。例如c1~4卤素烷基指氢原子被一个或多个卤素原子取代的包含1~4个碳原子的烷基。
81、“杂环”、“杂环烷基”、“杂环烷烃”指包含至少一个杂原子的饱和环或非芳香性的不饱和环;其中杂原子指氮原子、氧原子、硫原子;
82、“芳杂环”指包含至少一个杂原子的芳香性不饱和环;其中杂原子指氮原子、氧原子、硫原子;
83、“立体异构体”包括对映异构体和非对映异构体;
84、本发明的化合物可以包含不对称中心或手性中心,因此存在不同的立体异构体。本发明的化合物所有的立体异构形式,包括但绝不限于,非对映体,对映异构体,阻转异构体,和它们的混合物,如外消旋混合物,组成了本发明的一部分。很多有机化合物都以光学活性形式存在,即它们有能力旋转平面偏振光的平面。在描述光学活性化合物时,前缀d、l或r、s用来表示分子手性中心的绝对构型。这些立体异构体的化学结构是相同的,但是它们的立体结构不一样。特定的立体异构体可以是对映体,异构体的混合物通常称为对映异构体混合物。50:50的对映体混合物被称为外消旋混合物或外消旋体,这可能导致化学反应过程中没有立体选择性或立体定向性。术语“外消旋混合物”和“外消旋体”是指等摩尔的两个对映异构体的混合物,缺乏光学活性。
85、术语“药学上可接受的”是指某载体、运载物、稀释剂、辅料,和/或所形成的盐通常在化学上或物理上与构成某药物剂型的其它成分相兼容,并在生理上与受体相兼容。
86、本发明所述药物组合物,可以是任意一种可复用的药物制剂形式,如:口服,注射,外用等形式,口服剂型包括但不限于:片剂,胶囊剂,口服液,颗粒剂,丸剂,混悬剂,注射剂选自水针,粉针,外用制剂选自贴剂,膏剂。所有制剂均可以按照制剂学常规技术制备,如以本发明化合物,或其立体异构体、或其药学上可接受的盐中的任意一项作为药物活性成份,必要时加入药学上可接受的载体,制备成适于服用的上述药物剂型,其中,单位剂量的药物活性成份可以是0.1mg-1000mg,如片剂的每片含有0.1mg-1000mg优选5-500mg的药物活性成份。
87、术语“盐”和“可药用的盐”是指上述化合物或其立体异构体,与无机和/或有机酸和碱形成的酸式和/或碱式盐,也包括两性离子盐(内盐),还包括季铵盐,例如烷基铵盐。这些盐可以是在化合物的最后分离和纯化中直接得到。也可以是通过将上述化合物,或其立体异构体,与一定数量的酸或碱适当(例如等当量)进行混合而得到。这些盐可能在溶液中形成沉淀而以过滤方法收集,或在溶剂蒸发后回收而得到,或在水介质中反应后冷冻干燥制得。本发明中所述盐可以是化合物的盐酸盐、硫酸盐、枸橼酸盐、苯磺酸盐、氢溴酸盐、氢氟酸盐、磷酸盐、乙酸盐、丙酸盐、丁二酸盐、草酸盐、苹果酸盐、琥珀酸盐、富马酸盐、马来酸盐、酒石酸盐或三氟乙酸盐。
88、在某些实施方式中,本发明的一种或多种化合物可以彼此联合使用。也可选择将本发明的化合物与任何其它的活性试剂结合使用,用于制备调控细胞功能或治疗疾病的药物或药物组合物。如果使用的是一组化合物,则可将这些化合物同时、分别或有序地对受试对象进行给药。
89、对本发明的化合物的制备方法进行说明,具体步骤如下:
90、步骤1:
91、
92、式b、式c中rp表示氢原子或亚氨基的保护基团,作为rp的亚氨基的保护基团,优选为苄基、对甲氧基苄基、叔丁氧基羰基、苄氧基羰基等。式b、式c和式d中的r1基团参见上述的定义,与前文的定义相同。
93、在制备方法1中,使式a所示化合物与式b所示的肼衍生物在碱的存在下反应得到式c所示化合物,反应通常可以在三乙基胺、二异丙基乙基胺dipea、吡啶、4-二甲基氨基吡啶等有机碱或氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠等无机碱的存在下进行反应;例如可以在二氯甲烷、氯仿、四氢呋喃、乙醚、苯、甲苯、二甲苯、二甲基甲酰胺等或混合溶剂等惰性溶剂中进行。然后进行脱保护反应,通过该化合物环化形成式d所示的化合物。上述碱的用量相对于式a化合物1摩尔,优选为等摩尔~过量摩尔,更优选为1摩尔量-5摩尔量,最优选为1摩尔-3摩尔。此外,该碱为液体时,可以将该碱兼用作溶剂和碱。反应温度通常为-78℃-200℃,优选为20-100℃。反应时间通常为5分钟-7天,优选为8小时-96小时。
94、在制备方法1中,使式c化合物进行脱保护和环化反应得到式d所示的化合物,其中脱保护反应中反应试剂选自三氟乙酸、盐酸溶液等、溶剂选择甲醇、二氯甲烷或1,4-二氧六环等,优选tfa/ch2cl2的方式脱除保护基;如果使用boc作为保护基时,脱保护反应可以在标准条件下进行,例如,二氯甲烷/三氟乙酸体系、饱和的氯化氢二氧六环溶液中进行;其中所述的环化反应的反应条件为碱性条件,碱性条件选自一定浓度的氢氧化钠溶液、氢氧化钾溶液、碳酸钠溶液、碳酸钾溶液或碳酸氢钠溶液等,优选为氢氧化钠溶液。脱保护和环化反应温度通常为-78℃-200℃,优选为20-100℃,反应时间通常为5分钟-7天,优选为8小时-96小时。
95、步骤2:
96、式g所示的化合物可以通过步骤2-1或步骤2-2的方式制备得到,步骤2-1或步骤2-2的制备方法如下:
97、步骤2-1:
98、
99、式e和式g所述化合物的取代基r1和r2基团参见上述的定义,与前文的定义相同。该反应中通过c-n偶联反应将式e和式d化合物反应制备得到式g所示化合物。反应溶剂为1,4二氧六环、四氢呋喃、乙醚、苯、甲苯、二甲苯等或混合溶剂中进行,反应温度为0-200摄氏度,优选为20-150℃。
100、步骤2-2:
101、
102、式f和式g所述化合物的取代基r1和r2基团参见上述的定义,与前文的定义相同,卤原子为f、cl、br、i。式f所示化合物与式d所示化合物经c-n偶联反应得到式g所示化合物,反应溶剂为1,4二氧六环、四氢呋喃、乙醚、苯、甲苯、二甲苯等或混合溶剂中进行,反应温度为0-200摄氏度,优选为20-150℃。该步骤中所述c-n偶联反应为本领域惯常用于构建c-n键的偶联方法,例如ullmann反应,buchwald反应,优选ullmann反应,更优选碘化亚铜/碳酸钾/n,n-二异丙基乙胺dmeda/1,4-二氧六环的反应条件(偶联反应的条件为cui,dmeda,k2co3,1,4-dioxane),或更优选为cui/k2co3/n,n'-二甲基-1,2-环己二胺、苯甲醚/nai/微波,或更优选为cui/k2co3/苯甲醚/nai/微波。
103、步骤2-1和2-2中化合物r2b(oh)2和r2-卤原子可以通过有机化学领域中通常的合成方法以简单易得的原料制备得到。
104、步骤3:
105、
106、式h和式g所述化合物的取代基r1、r2、r3、r4基团参见上述的定义,与前文的定义相同。式g所示化合物先在氧化剂的作用下形成活性较高的中间体亚砜,然后再与式h所示化合物进行取代反应得到式i所示化合物。其中反应溶剂选自二氯甲烷、氯仿、四氢呋喃、乙醚、苯、甲苯、二甲苯、二甲基甲酰胺等或其混合溶剂;在氧化反应中所述氧化剂优选为间氯过氧苯甲酸m-cpba;所述取代反应的条件为本领域惯常用于取代的反应条件,例如碱性条件或酸性条件,碱性条件优选二异丙基乙胺dipea,酸性条件优选三氟乙酸,反应温度为-20-200℃,优选为20-150℃,最优选为室温。
107、在步骤3中式h所示取代苯胺类化合物可以通过有机化学领域中通常的合成方法以简单易得的原料制备得到。
108、步骤4:如若步骤3中制备得到式i化合物含有手性中心,本领域技术人员可结合已知的分离技术通过色谱方法或其他拆分方法得到纯手性化合物。例如可通过sfc拆分得到含有一个手性中心的两个手性化合物。如果步骤3中得到的式i化合物不含有手性中心,就不需要进行步骤4的拆分过程。
109、显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
110、以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
- 还没有人留言评论。精彩留言会获得点赞!