一种FGFR激酶抑制剂及其制药用途
本发明属于药物开发领域,具体涉及一种fgfr激酶抑制剂及其制药用途。
背景技术:
1、肿瘤分子靶向治疗通过利用肿瘤组织或细胞所具有的特异性结构分子为靶点,使用能与这些靶分子特异性结合的药物,特异性地杀伤肿瘤细胞。以激酶抑制剂为代表的肿瘤靶向药物由于具有药效迅速、毒副作用轻微等优势而使近10年来恶性肿瘤的治疗发生了革命性变化。
2、成纤维细胞生长因子(fibroblast growth factor,fgf)是一种有丝分裂信号分子,包含20多种蛋白,在许多组织中对控制细胞增殖、分化和存活起重要作用。fgf的作用高度依赖于它们对特定受体的亲和力,成纤维细胞生长因子受体(fibroblast growthfactor receptor,fgfr)是fgf的主要受体,其本质是受体型蛋白酪氨酸激酶(tyrosinekinase,tk),目前已知的fgfr主要包括4种类型,即fgfr1、fgfr2、fgfr3及fgfr4。研究表明通过靶向抑制fgfr的异常表达可实现对肿瘤的控制。据报道,fgfr1的过表达会引发乳腺癌和鳞状非小细胞肺癌等疾病;fgfr2突变会导致胃癌和子宫内膜癌等病症;fgfr3异常表达与尿路上皮癌等相关;fgfr4的过表达可以增加胃癌、肝癌、前列腺癌、横纹肌肉瘤、卵巢癌、结肠癌、皮肤黑素瘤等的发病概率。
3、
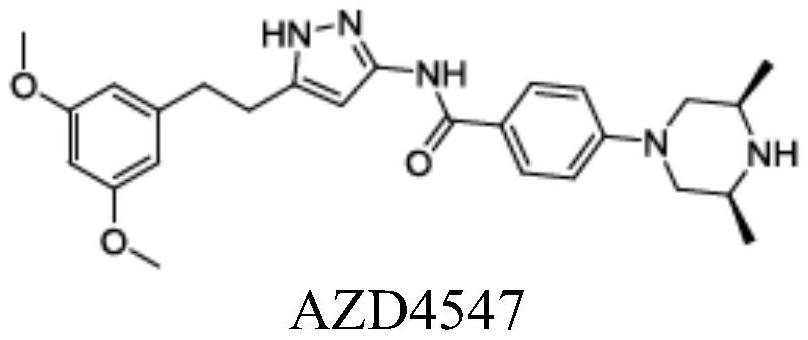
4、成纤维细胞生长因子受体4(fgfr4)是fgfr家族中较晚被发现的一类跨膜酪氨酸激酶受体。fgfr4基因的基因组织包含18个外显子,其igⅲ结构域一半的c末端不像fgfr1-3那样在三种替代形式之间变化。目前针对fgfr1-3抑制剂取得了较多研究成果,但针对特异性fgfr4抑制剂的研究报道则相对较少。许多选择性fgfr抑制剂最近已开始临床开发,如ap24534,bgj398,azd4547,ly2874455和jnj-42756493。这些化合物对fgfr1,fgfr2和fgfr3的体外激酶活性非常高,但对fgfr4的抑制作用不是很理想。例如,paul r.gavined等设计的n-[5-[2-(3,5-二甲氧基苯基)乙基]-2h-吡唑-3-基]-4-(3,5-二乙基哌嗪-1-基)苯甲酰胺(azd4547)对乳腺癌和骨髓癌有一定的疗效。在细胞中,azd4547通过frs2,plcγ和mapk有效抑制fgfr1、2和3酪氨酸激酶的自身磷酸化,但对fgfr4细胞激酶活性抑制较弱。开发出对fgfr4具有优异的抑制活性的药物具有重要意义。
5、另外,第一代fgfr4抑制剂或一些多靶点抑制剂(如索拉非尼等),在经过9~12个月的临床用药后通常会发生明显的继发性耐药(如v550l残基错位突变),极大限制了肿瘤患者生存时间的延长(gao l,wang x,tang y,et al.fgf19/fgfr4signaling contributesto the resistance of hepatocellular carcinoma tosorafenib[j].j expclin cancerres,2017,36(1):8.)。但截至目前,可用于克服fgfr4v550l突变的小分子抑制剂还鲜有报道。
技术实现思路
1、本发明的目的在于提供一种fgfr激酶抑制剂及其制药用途。
2、本发明提供了式i所示化合物或其盐、立体异构体、氘代化合物:
3、
4、其中,a环为5~6元饱和环烷基、5~6元饱和杂环基、5~6元芳基或5~6元杂芳基;
5、m为0~5的整数;
6、r1各自独立地选自未取代或被r5取代的以下基团:c1~6烷基、c1~6烷氧基;r5为羟基、氨基、卤素、羧基、氰基、c1~6烷氧基或c1~6烷基;r6为c1~6烷基;
7、r2为卤素、c1~6烷基或c1~6烷氧基;
8、r3为卤素、c1~6烷基或c1~6烷氧基;
9、r4为卤素、c1~6烷基或c1~6烷氧基;
10、为或-。
11、进一步地,a环为5~6元饱和环烷基或5~6元饱和杂环基;
12、m为0~3的整数;
13、r1各自独立地选自未取代或被r5取代的以下基团:c1~4烷基、c1~4烷氧基;r5为羟基、氨基、卤素、羧基、氰基、c1~4烷氧基或c1~4烷基;r6为c1~4烷基;
14、r2为卤素;
15、r3为卤素;
16、r4为c1~4烷基。
17、进一步地,所述化合物的结构如式ii所示:
18、
19、其中,a环为5~6元饱和杂环基;
20、m为0~2的整数;
21、r1各自独立地选自未取代或被r5取代的c1~4烷基;r5为羟基、c1~4烷氧基或c1~4烷基;r6为c1~4烷基。
22、进一步地,所述化合物的结构如式iii-1、式iii-2或式iii-3所示:
23、
24、其中,r1选自未取代或被r5取代的c1~2烷基;r5为羟基、c1~2烷氧基或c1~2烷基;r6为c1~2烷基。
25、进一步地,所述化合物的结构如式iv所示:
26、
27、其中,a环为5~6元饱和杂环基;
28、m为0~2的整数;
29、r1各自独立地选自未取代或被r5取代的c1~4烷基;r5为羟基、c1~4烷氧基或c1~4烷基;r6为c1~4烷基。
30、进一步地,所述化合物的结构如式v-1、式v-2或式v-3所示:
31、
32、其中,r1选自未取代或被r5取代的c1~2烷基;r5为羟基、c1~2烷氧基或c1~2烷基,r6为c1~2烷基。
33、进一步地,所述化合物选自:
34、
35、
36、
37、本发明还提供了一种治疗癌症的药物,它是以上述化合物或其盐、立体异构体、氘代化合物为活性成分,加上药学上可接受的辅料制备而成的制剂。
38、本发明还提供了上述化合物或其盐、立体异构体、氘代化合物在制备fgfr激酶抑制剂中的用途。
39、进一步地,所述fgfr激酶抑制剂为fgfr4激酶抑制剂。
40、进一步地,所述fgfr4激酶抑制剂为预防和/或治疗与fgfr4激酶活性有关的疾病的药物。
41、进一步地,所述fgfr激酶抑制剂为fgfr4v550l突变体抑制剂。
42、进一步地,所述fgfr4v550l突变体抑制剂为预防和/或治疗与fgfr4v550l突变有关的疾病的药物。
43、进一步地,所述疾病为胃癌、肝癌、前列腺癌、横纹肌肉瘤、卵巢癌、结肠癌或皮肤黑素瘤。
44、实验结果表明,本发明化合物对fgfr4激酶具有优异的抑制效果,可以作为fgfr4激酶抑制剂。
45、本发明化合物可以作为fgfr4野生型和突变体的抑制剂。
46、本发明化合物能够有效抑制ba/f3-etv6-fgfr4-v550l细胞增殖,有效克服fgfr4v550l突变,可以作为fgfr4v550l突变体抑制剂。
47、本领域技术人员公知的,抑制fgfr4激酶活性能够有效预防和治疗与fgfr4激酶活性有关的肿瘤(包括胃癌、肝癌、前列腺癌、横纹肌肉瘤、卵巢癌、结肠癌、皮肤黑素瘤等)。
48、本发明化合物能够用于制备预防和/或治疗与fgfr4激酶活性有关,以及与fgfr4v550l突变有关的疾病的药物,应用前景广阔。
49、显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
50、以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
- 还没有人留言评论。精彩留言会获得点赞!