一种磺达肝癸钠二糖中间体的制备方法与流程
本发明涉及医药,尤其涉及一种磺达肝癸钠二糖中间体甲基4-o-[2-o-乙酰基-6-甲基-3-o-苄基-β-l-吡喃艾杜糖醛酸基]-2-脱氧-2-[[苄氧羰基]氨基]-3-o-苄基-α-d-吡喃葡萄糖苷6-乙酸酯的制备方法。
背景技术:
1、磺达肝癸钠由赛诺菲圣德拉堡集团和欧加农公司(后并入默沙东)联合原研,在美国最早于2001年(mylan公司)获得fda批准上市,用于术后深静脉血栓的预防。2002年,获批用于肺栓塞、血栓栓塞的治疗。2004年,赛诺菲与安万特合并时,将其授权转让给葛兰素史克公司(gsk)。2007年磺达肝癸钠获批新适应症急性冠状动脉综合征的治疗。2008年,磺达肝癸钠进入中国市场,商品名为安卓(arixtra)。
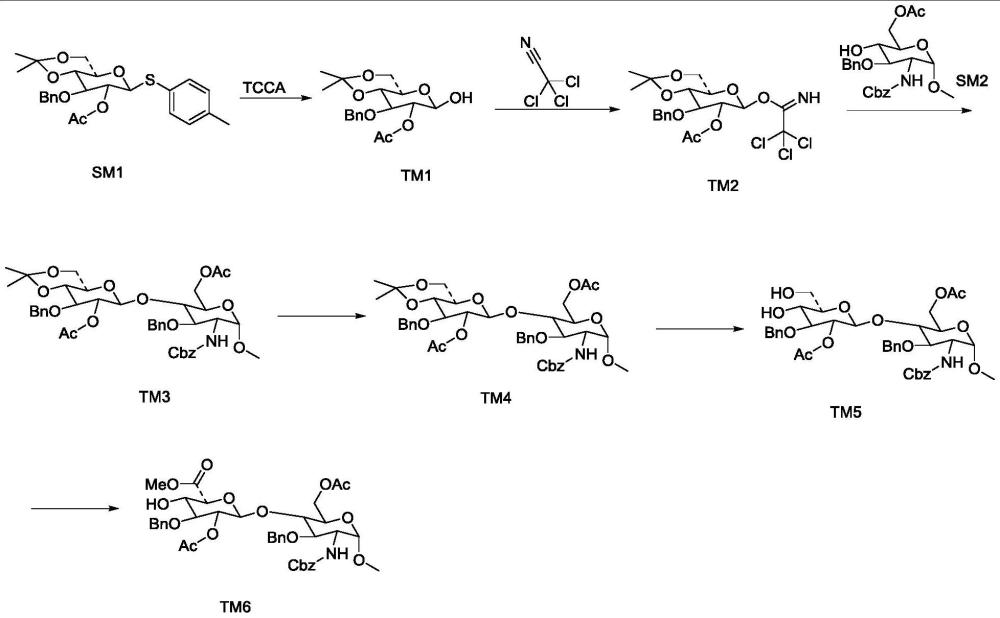
2、磺达肝癸钠和传统肝素药品相比具有众多优势,比如:1)完全化学合成,质量标准可控制,单一的化学实体成分,无病菌污染风险;2)高度的靶点选择性,它是xa凝血因子选择性抑制剂,基本上不会导致血小板减少和出血的副作用;3)更长的半衰期(17个小时以上)以及更高的生物利用度。总之,作为肝素类抗凝血药的顶级产品,由于其优异的治疗效果和较强的市场潜力,磺达肝癸钠长期受到药物化学家和合成化学家们的关注。但在磺达肝癸钠的制备合成研究中,磺达肝癸钠二糖中间体关键杂质即化合物ⅶ甲基4-o-[2-o-乙酰基-6-甲基-3-o-苄基-β-l-吡喃艾杜糖醛酸基]-2-脱氧-2-[[苄氧羰基]氨基]-3-o-苄基-α-d-吡喃葡萄糖苷6-乙酸酯的质量尤为重要。现有磺达肝癸钠二糖中间体的合成工艺路线如下:
3、
4、该路线的不足之处:1、合成路线长,且tm1为半缩醛结构、tm2中间体为活性亚胺酯极不稳定,放置过程中会缓慢变质,难以存放。2、步骤三中,催化剂筛选了tmsotf、tbsotf、agotf其中一种,溶剂筛选了二氯甲烷、甲苯、四氢呋喃、乙腈等,均无法实现,均未检测到β异构体的产生(1,2-顺式糖苷键的生成),无法得到化合物ⅱβ,也就无法合成该杂质。3、步骤二中使用到的三氯乙腈属于3类致癌物,对人体健康以及环境污染较大。因此,目前急需对其进行工艺改进。
技术实现思路
1、基于目前技术现状,本发明目的在于提供一种磺达肝癸钠二糖中间体杂质甲基4-o-[2-o-乙酰基-6-甲基-3-o-苄基-β-l-吡喃艾杜糖醛酸基]-2-脱氧-2-[[苄氧羰基]氨基]-3-o-苄基-α-d-吡喃葡萄糖苷6-乙酸酯的制备方法,实现高收率、简单、绿色合成。
2、其合成路线如下:
3、
4、具体包括如下步骤:
5、s1:化合物sm1上tbs保护得到化合物ⅰ;
6、s2:化合物ⅰ与化合物sm2进行糖苷化,同时生成了化合物ⅱα和ⅱβ,经柱层析分离得到化合物ⅱβ;
7、s3:化合物ⅱβ与吡啶氢氟酸盐反应脱除tbs保护得到化合物ⅲ;
8、s4:化合物ⅲ与乙酰化试剂反应得到化合物ⅳ;
9、s5:化合物ⅳ脱除丙酮叉保护基得到化合物ⅴ;
10、s6:化合物ⅴ经过tempo/碘苯二乙酸选择性氧化得到化合物ⅵ;
11、s7:化合物ⅵ经过甲基化成酯得到化合物ⅶ;
12、优选:
13、s1中,sm1在2,6-二甲基吡啶、叔丁基二甲硅基三氟甲磺酸酯体系中反应,化合物sm1与2,6-二甲基吡啶、叔丁基二甲硅基三氟甲磺酸酯的摩尔比为1:1.5~2.5:1.2~2,反应溶剂为二氯甲烷、四氢呋喃、1,4-二氧六环其中一种,反应温度范围-10~30℃,时间为3~5h。
14、s2中,选用tmsotf、tbsotf、agotf其中一种为催化剂,与化合物sm2摩尔比为0.1~0.4:1,n-碘代丁二酰亚胺为助剂,与化合物sm2摩尔比为1.5~2:1,加入的干燥剂为新活化的4a分子筛,与化合物sm2质量比为1.5~2.5:1,反应溶剂为二氯甲烷、四氢呋喃、甲苯其中的一种,反应温度范围-40~-10℃,时间为1~3h。
15、s3中,以质量百分比30%吡啶氢氟酸溶液为反应试剂,与化合物ⅱβ的摩尔比为4~10:1,反应溶剂为吡啶、二氯甲烷、四氢呋喃其中的一种,反应温度范围0~25℃,时间为2~4h。
16、s4中,乙酰化试剂为乙酸酐或乙酰氯其中的一种,与化合物ⅱβ的摩尔比为1.3~1.8:1;以4-二甲氨基吡啶为催化剂,与化合物ⅱβ的摩尔比为0.1~0.3:1;反应溶剂为吡啶、二甲基吡啶、四氢呋喃的一种,反应温度范围60~80℃,时间为12~16h。
17、s5中,反应溶剂为冰乙酸与水的混合溶剂,其体积比为6:4~8:2,反应温度范围20~30℃,时间为4~16h。
18、s6中,tempo为氧化剂,碘苯二乙酸为助剂,化合物ⅴ、tempo及碘苯二乙酸的摩尔比为1:0.2~0.4:2~4,反应溶剂为二氯甲烷、1,2-二氯乙烷、甲苯其中一种与水的混合溶剂,其体积比为3:1~1:1,反应温度范围25~30℃,时间为2~8h。
19、s7中,在碱性条件下进行甲基化反应,所述碱为碳酸钾,其与化合物ⅵ的摩尔比为1.5~2:1;所述甲基化试剂选自碘甲烷或硫酸二甲酯,其与化合物ⅵ的摩尔比为1.2~2:1,反应溶剂为n,n-二甲基甲酰胺、四氢呋喃、乙腈其中一种,反应温度范围25~40℃,时间为2~4h。
20、相比与之前的二糖中间体合成工艺而言,本发明的关键在于:步骤⑵通过在化合物sm1的c2位引入tbs,减少s2步骤糖苷化过程中的邻位参与效应,从而巧妙地改变了糖苷化的结果。相较于传统的只得到α构型产物的合成路线,本发明可以得到1:1比例的α、β两种构型的二糖混合物,然后,经柱层析分离,得到β构型的手性关键中间体化合物iiβ。在步骤⑶中,尝试了tbaf,hcl等脱除tbs的条件,结果会导致糖苷键的断裂氟化取代或糖环上乙酰基的水解,故选用更温和的脱除条件吡啶氢氟酸盐来实现,不仅选择性好且收率高易于处理,步骤(6)中,相比于传统工艺tempo/次氯酸钠溶液,乙腈为溶剂体系,该工艺采用tempo为氧化剂,碘苯二乙酸为助剂,采用二氯甲烷/水两相体系,实现化合物ⅴ的伯醇选择性氧化,反应高效,选择性好转化率高,后处理硫代硫酸钠洗涤二氯甲烷相,浓缩即可得到化合物ⅵ,后处理更简单、绿色,进而完成该关键杂质的制备合成。总收率达44.88%以上,实现了该中间体的高收率、简单、绿色合成。
技术特征:
1.一种磺达肝癸钠二糖中间体的制备方法,其特征在于,通过如下步骤实现:
2.根据权利要求1所述磺达肝癸钠二糖中间体的制备方法,其特征在于,s1中,化合物sm1在2,6-二甲基吡啶、叔丁基二甲硅基三氟甲磺酸酯体系中反应;
技术总结
本发明属医药技术领域,公开了一种磺达肝癸钠二糖中间体的制备方法,涉及磺达肝癸钠二糖中间体甲基4‑O‑[2‑O‑乙酰基‑6‑甲基‑3‑O‑苄基‑β‑L‑吡喃艾杜糖醛酸基]‑2‑脱氧‑2‑[[苄氧羰基]氨基]‑3‑O‑苄基‑α‑D‑吡喃葡萄糖苷6‑乙酸酯的制备。其合成路线如下:该方法通过在化合物SM1的C2位引入TBS,减少S2步骤糖苷化过程中的邻位参与效应,改变了糖苷化中异构体的生成结果。步骤⑸采用TEMPO为氧化剂,碘苯二乙酸为助剂,采用二氯甲烷/水两相体系,实现化合物Ⅴ的伯醇选择性氧化,反应高效,选择性好转化率高。总收率达44.88%以上,实现了该中间体的高收率、简单、绿色合成。
技术研发人员:王海军,范琪琦,王鹏,张凤,崔浩
受保护的技术使用者:新天地医药技术研究院(郑州)有限公司
技术研发日:
技术公布日:2024/3/11
- 还没有人留言评论。精彩留言会获得点赞!