一种水溶性纳米发光材料及其制备方法与流程
本发明属于纳米材料技术领域,涉及一种水溶性纳米发光材料、制备方法及应用。
背景技术:
纳米发光材料由于其优越的发光新能及其在化学传感器、生物标记以及医学成像等领域的广泛应用,逐渐成为新功能材料中重要的一个分支。纳米发光材料是指基质的粒子在1~100nm的发光材料。
纳米发光材料在有机溶剂中的光转化效率均较高,而在水溶液中却会有很大的衰减,导致了在相同x射线剂量的激发下,水溶性发光中心的光产额显著低于油溶发光中心。而在生物标记领域中,不能使用有机溶剂与机体直接接触,所以必须增强纳米发光中心在水溶液中的溶解性,以便更好地应用于机体。
目前已有多种对纳米发光材料进行水溶性修饰的方法,如以硅为水溶性外壳,虽然大大提高了纳米发光中心在水中的溶解性,但却显著降低了纳米发光颗粒的光产额,使其生物应用受限。所以如何更好地对纳米发光中心进行水溶性修饰、更大程度地保留其光转化效率便成了研究的焦点。
技术实现要素:
针对上述技术问题,本发明提供了一种纳米发光材料亲水性修饰的方法,解决现有的水溶性修饰方式显著降低了纳米发光颗粒的光产额,使其生物应用受限的问题。
为了实现上述目的,本发明采用如下技术方案予以实现:
一种水溶性纳米发光材料的制备方法,包括以下步骤:
在纳米发光颗粒表面包覆油酸或油氨形成油溶性纳米发光材料,将油溶性纳米发光材料加入环己烷溶液中形成纳米发光材料的环己烷溶液;
将氨基咪唑溶于丙酮和水的混合液中,得到氨基咪唑离子溶液;
将氨基咪唑离子溶液与纳米发光材料的环己烷溶液按照纳米发光材料的质量:氨基咪唑的质量为20:1~2:1混合;室温下反应4~24h,所得沉淀即为水溶性纳米发光材料。
具体的,所述的纳米发光材料为氟化镧纳米发光材料。
具体的,所述的丙酮和水按照体积比为2:1的比例混合。
具体的,所述的纳米发光材料的环己烷溶液中纳米发光材料的浓度为1~10mg/ml,氨基离子液体的浓度为1ug/ml~1mg/ml。
本发明还公开了一种上述制备方法获得到的水溶性纳米发光材料。
与现有技术相比,本发明的有益效果是:
本发明使用氨基咪唑离子液体修饰纳米发光粒子,所得的水溶性纳米发光材料在水中均呈现出良好的稳定性,光谱学分析结果表明,水溶性纳米发光颗粒均保持了极高的光产额,与油溶性的纳米发光颗粒相比,光产额损失在10%之内,为后续的成像及治疗实验提供了生物相容性好的发光材料基础。
附图说明
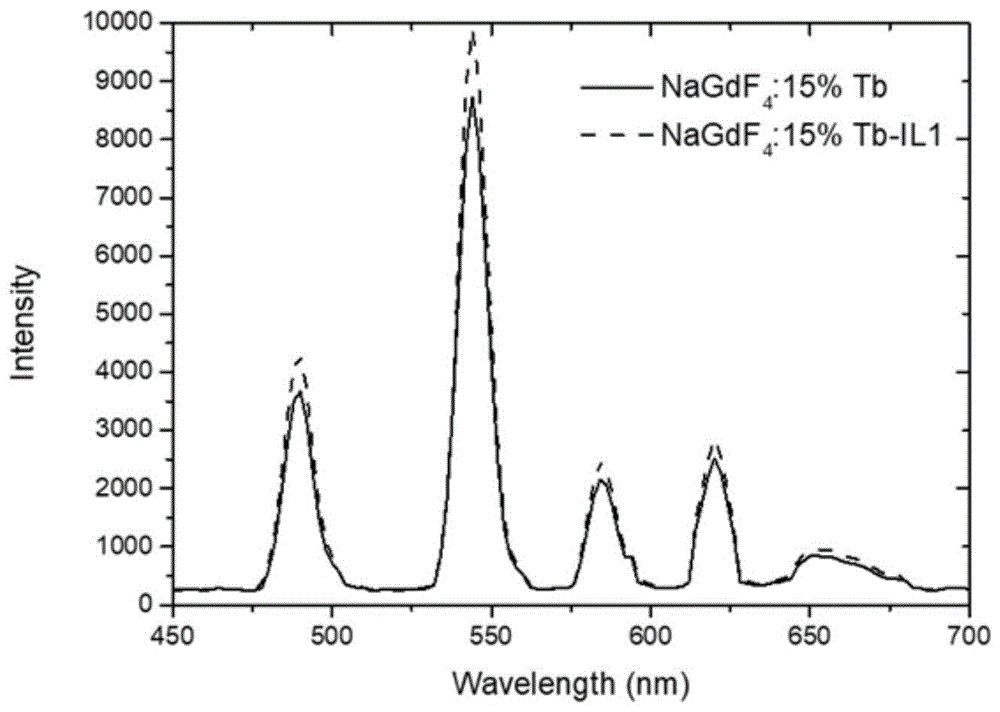
图1是实施例1中nagdf4:15%tb纳米发光材料经氨基咪唑离子液体修饰前后光谱对比图;
图2是实施例2naluf4:15%tb纳米发光材料经氨基咪唑离子液体修饰前后光谱对比图;
图3是对比例1中nagdf4:15%tb纳米发光材料经peg修饰前后光谱对比图;
图4是对比例2中naluf4:15%tb纳米发光材料经peg修饰前后光谱对比图。
图5是nagdf4:15%tb-il1-mc540水溶性纳米发光颗粒(1mg/ml)在不同x射线剂量下的发光强度图;
图6是为不同浓度nagdf4:15%tb-il1-mc540水溶性纳米发光颗粒在x射线剂量为1.0gy下的发光强度图;
图7为naluf4:15%tb-il1-mc540水溶性纳米发光颗粒(1mg/ml)在不同x射线剂量下的发光强度图;
图8为不同浓度naluf4:15%tb-il1-mc540水溶性纳米发光颗粒在x射线剂量为0.1gy下的发光强度图。
具体实施方式
本发明公开的一种水溶性纳米发光材料的制备方法,包括以下步骤:
步骤1:在纳米发光材料的表面包覆有油酸或油氨形成油溶性纳米发光材料,再将其加入环己烷溶液中形成油溶性纳米发光材料的环己烷溶液,本发明优选的,纳米发光材料的环己烷溶液中纳米发光材料的浓度为1~10mg/ml;本发明中的纳米发光材料为氟化镧纳米发光材料,如下述实施例所列举的nagdf4:15%tb或naluf4:15%tb。
将氨基咪唑离子液体溶于丙酮和水的混合液中,得到氨基咪唑离子溶液;本发明优选的,丙酮和水按照体积比为2:1的比例混合;本发明优选的,氨基离子液体的浓度为1ug/ml~1mg/ml。
本发明所使用的氨基咪唑离子液体(以下简称“il1”)可通过市面购买,其结构为:
步骤2:将氨基咪唑离子液体与纳米发光材料的环己烷溶液按照纳米发光材料的质量:氨基咪唑的质量为20:1~2:1混合;室温下反应4~24h,所得沉淀即为水溶性纳米发光材料。
氨基咪唑离子附着在纳米发光颗粒表面形成水溶性纳米发光材料。将该纳米发光材料与光敏剂mc540耦合,构建的耦合系统具有低剂量激发的高单态氧产生量,为实现低剂量x射线激发光动力学治疗提供了材料基础。
在本发明中,油溶的纳米发光材料表面包覆油酸,为负电荷羧基表面,可与离子液体咪唑环的正电荷作用,通过静电作用将离子液体包覆于纳米发光材料表面,赋予其水溶性。同时,在水溶液中,氨基质子化产生的正电荷也可与油酸羧基负离子进行静电作用,使离子液体与纳米发光材料之间结合的更紧密。由于所选用的氨基咪唑离子液体分子量小,且与纳米发光材料间结合紧密,且形成的水化层较小,因此对纳米发光材料的光产额具有极大的保护作用。
以下给出本发明的具体实施例,需要说明的是本发明并不局限于以下具体实施例中,凡在本申请技术方案基础上做的等同变换均落入本发明的保护范围。
实施例1
步骤1,对油溶的nagdf4:15%tb纳米发光材料进行定容至10ml,取0.5ml用差量法进行干燥称重,计算整体溶液中纳米发光材料的浓度,以mg/ml为单位。配制10mg/ml的纳米发光材料环己烷溶液;
本实施例采取的氨基离子液体为氨基咪唑离子溶液,称取氨基离子液体il1,溶于丙酮:水按照体积比为2:1的溶液中,制备1mg/ml的il1溶液。
步骤2,将il1溶液与纳米发光材料的环己烷溶液(10mg/ml)以体积比1:2混合(即纳米发光材料与氨基咪唑的质量比为20:1),室温下磁力搅拌16小时。反应完全后,在12000rpm离心混合液20分钟),弃去上清。以4ml水分散沉淀,洗涤三遍,得到水溶性的纳米发光颗粒。
本实施例制备的水溶性的纳米发光颗粒的光谱图如图1所示。纳米粒子浓度为1mg/ml,x射线照射剂量为80kev,0.5ma。
将本实施例得到的水溶性纳米发光颗粒与光敏剂mc540耦合,构建了nagdf4:15%tb-il1-mc540耦合系统,在不同x射线激发条件下即不同耦合系统浓度以sosg法,通过sosg-ep的荧光强度考察单态氧产生率,结果如图5所示。
实施例2
本实施例与实施例1的区别在于:纳米发光材料为naluf4:15%tb。
本实施例制备的水溶性的纳米发光颗粒的光谱图如图2所示。
将本实施例得到的水溶性纳米发光颗粒与光敏剂mc540耦合,构建了naluf4:15%tb-il1-mc540耦合系统,在不同x射线激发条件下即不同耦合系统浓度以sosg法,通过sosg-ep的荧光强度考察单态氧产生率。结果如图2所示。
实施例3
本实施例与实施例1的区别是:纳米发光材料与氨基咪唑的质量比为2:1。将本实施例制备的水溶性的纳米发光颗粒与光敏剂mc540耦合,构建了nagdf4:15%tb-il1-mc540耦合系统,在不同x射线激发条件下即不同耦合系统浓度以sosg法,通过sosg-ep的荧光强度考察单态氧产生率,发现结果略低于实施例1,但仍较现有的修饰方法高。
对比例1
本对比例采用peg水溶性修饰方法对纳米发光颗粒进行修饰,具体方法为:
步骤1,对油溶的nagdf4:15%tb纳米发光材料进行定容至5ml,取0.3ml用差量法进行干燥称重,计算整体溶液中纳米发光材料的浓度,以mg/ml为单位;
步骤2,将溶剂完全干燥后加入约5ml二氯甲烷(dcm),并加入搅拌磁子;
步骤3,按纳米发光材料与peg-nh2质量比1:6称取peg,并溶于约5mldcm中;
步骤4,分别在步骤2和步骤3的溶液液面上方通氩气1min,以排除容器内的空气;
步骤5,将步骤3的溶液迅速倒入步骤2的溶液中,随后用氩气通入液面以下保持5min,以排除溶液内部的空气;迅速将容器封口,以200rpm搅拌48h;
步骤6,将溶液干燥,加入6ml去离子水,12000rpm离心30min,弃上清,将沉淀溶于10ml去离子水,即得到peg修饰的nagdf4:15%tb。
本对比例制备的水溶性的纳米发光颗粒的光谱图如图3所示。
对比例2
本对比例与对比例1的区别在于:纳米发光材料为naluf4:15%tb。
本对比例制备的水溶性的纳米发光颗粒的光谱图如图4所示。
综合实施例1~2和对比例1~2发现,以peg水溶性修饰方法作为参考,如图1所示,本发明氨基咪唑离子液体为水溶性修饰配基所制备的水溶性纳米发光颗粒,光产额较peg修饰的显著提高。
如图5所示,将氨基咪唑离子液体修饰后的纳米发光材料与光敏剂mc540耦合,构建的耦合系统具有低剂量激发的高单态氧产生量,为实现低剂量x射线激发光动力学治疗提供了材料基础。
- 还没有人留言评论。精彩留言会获得点赞!