用于生物气和生物油的共同生产的废水污泥和木质纤维素生物质的水热液化协同处理的制作方法
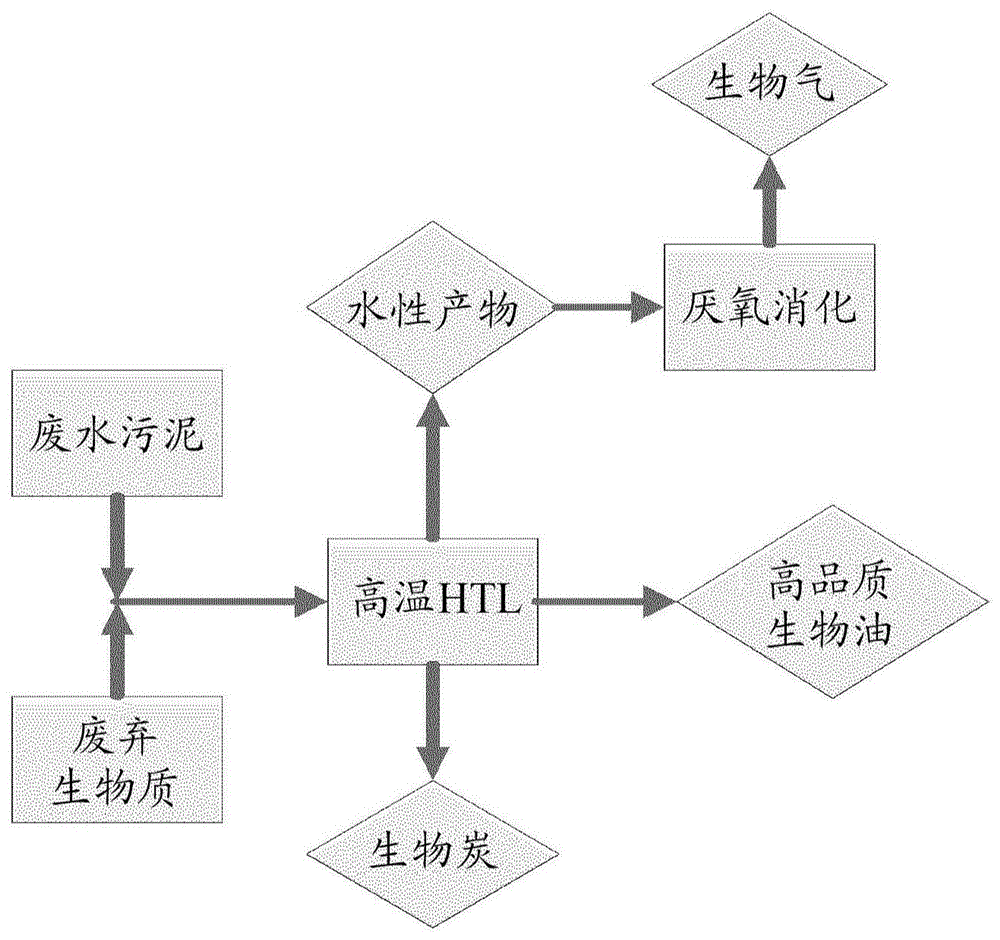
本公开涉及使用水热液化(htl)处理从高含水量废水污泥和木质纤维素生物质中共同生产生物气和高质量生物原油。
背景技术:
由于化石燃料储备的收缩和气候变化问题,人们对可再生能源的兴趣日益增长,导致对诸如生物质和废物的可再生能源生产的气态和液态燃料进行了广泛的研究。来自城市和工业废物如废水污泥的能量产生也是处理大量废物处理的环境友好方式,其另外的优点是消除来自能源作物衍生的生物燃料的部分间接温室气体排放[1]。由于废水的生物处理,城市和工业废水处理厂产生大量废活性污泥(was)。这种产生的污泥对环境构成威胁,在处理或焚烧之前需要进一步处理[2]。污泥处理和管理成本可能高达废水处理过程总成本的25-50%[3]。最近,人们越来越关注开发更环保的工艺以减少用于处理的污泥量,并通过将污泥转化为生物能来取代传统的污泥处理方法,例如填埋处理和焚烧。
改进的生物固体管理已被确定为城市污水排放管理的全加拿大战略的目标研究领域。得到加拿大环境部长理事会的赞同,它与对环境友好型工艺的兴趣不断增加相一致,以减少污泥处理量,并找到利用该物质生产生物能源和更有价值产品的方法。
全球正在开发无数的立即使用的生物燃料技术。在商业规模上操作的最先进的方法通常需要相对清洁、干燥和均匀的原料,例如初榨植物油、海藻油、废弃动物脂肪和用过的食用油。纳斯特石油公司(nesteoil)是将植物油加氢处理成烃类液体燃料的全球领导者,其商业规模工厂在芬兰、荷兰和新加坡运营,总生产能力为约每年200万吨。使用木质生物质作为原料,热解技术也正在商业化。初始产品,通常称为热解油或生物油,可用作低级加热油或可升级为工业标准烃类液体燃料。kior在密西西比州有一家商业工厂,每年生产40,000吨汽油、柴油和取暖油。霍尼韦尔公司的envergent技术使用ensyn的快速热处理
水热液化(htl)是一种热化学解聚过程,用于将湿生物质转化为粗制油,有时被称为生物油或生物原油,在中等温度和高压下发展为从生物质产生能量,其在存在水的情况下以避免能量密集型预先干燥[4]。它是一种很有前景的技术,在150-450℃的缺乏氧气的情况下和高达25-30mpa的压力下,用于将含水量高的废弃生物质转化为具有附加值的产品,主要是生物原油和固体残留物(生物炭)[5]。它消除了在其他热/热化学过程中所需要的昂贵的脱水/干燥过程的需要。水的显著特性如低介电常数和高离子产物在液化中起着重要的溶剂作用。使用一部分产出的油和炭可以使该过程在能量上自给自足,从而为htl过程提供热量。
该反应通常使用均相和/或非均相催化剂来改善所生产产物的质量和产率。有机起始原料(例如但不限于生物质、低等级煤(褐煤)和泥煤)的碳和氢被热化学转化成具有低粘度和高溶解度的疏水化合物。根据处理条件,所得到的燃料可以原样用于重型发动机,例如铁路或船用发动机,或者产出可以升级为运输燃料,包括喷气燃料、柴油和普通汽油。
htl技术为新兴快速热解过程提供了多种优势。虽然htl的工艺操作压力较高,但较低的温度和利用湿污泥的能力是关键优势。已发现,与焚烧相比,它具有成本效益[6],并且可以实现减少病原体的额外益处,同时满足对污泥土地应用的严格监管。此外,生产的生物油的质量较高,含水量较低(5%),氧含量较低(20-30%),且能量含量或热值较高(30-35mj/kg)。通过利用湿有机废物固体,我们的htl技术将代表生物燃料工业的重大进步,主要是通过利用现有的高水分有机废物的能力。
目前,在基于水解和水热处理的废水污泥中,仅建立或正在开发用于能量回收的单一的污泥到油的技术。kranich和eralp对污水污泥液化进行了早期研究[7]。在作为还原气体的氢气和催化剂如na2co3、nico3和na2mno4的存在下,污水污泥在不同的反应温度下转化为油。用水作为反应介质,油产率小于20wt%[7],[8]。molton等人进行了中试规模研究,其中具有20%总固体(ts)的初级和未消化污泥在连续反应器中在300℃和10mpa压力下加热,流速为30l/h,且液压停留时间为90分钟。该技术作为污泥到油反应系统(stors)获得了专利,油产率为10-20wt%,且炭为5-30wt%[5],[6]。它于2005年由thermoenergy公司商业化;然而,目前还没有全面安装运行。
污泥处理的另一个竞争过程是厌氧消化和沼气生产,并且有两个商业过程在进行。cambi工艺由三个容器(制浆容器、水解反应器和闪蒸罐)组成,并于160-180℃的温度在压力下处理污泥。cambi装置现在在挪威、丹麦、英格兰、爱尔兰、苏格兰和波兰进行运营。该技术相对复杂:在处理之前必须将来自废水处理的固体脱水至16%干固体,并且需要中压蒸汽供应。气味问题的报告与该过程相关[9]。
污泥消化(biothelys)工艺用于处理固体浓度高于10%的污泥,并于150-180℃和8-10巴(bar)的压力下进行操作。自1998年以来,法国一直在运营两个全尺寸设施。与cambi工艺一样,污泥消化(biothelys)工艺也可能受到气味问题的影响[9]。
发明概述
本文公开了由废活性污泥(was)和木质纤维素生物质的组合作为共进料的生物原油和生物气体生产。由于was具有高水百分比(>90%),添加木质纤维素生物质以增加固体浓度并提高废水液化的经济性。使用中心复合设计(ccd)方法优化操作条件如温度、反应时间和固体浓度。基于作者先前进行的催化剂筛选研究[10],将氢氧化钾(koh)在该过程中用作均相催化剂。废活性污泥和木质纤维素生物质如桦木和橡胶木屑/玉米秸秆/msw的混合物在作为均相催化剂的koh存在下在htl条件下进行转化。基于最大生物原油产量的响应面方法优化操作条件,包括反应温度、反应时间和固体浓度。通过将废活性污泥与木质纤维素生物质共进料,在310℃的最佳温度,10分钟的反应时间和10wt%的固体浓度下获得约34wt%的最高生物原油产率。该生物油与先前由锯屑在相同操作条件下产生的生物油的比较显示出生物原油的分子量显著改善,表明存在较轻的组分。生物原油产品的综合表征表明,这些生物油具有较低的热稳定性、较高的挥发性物质和较低的固定碳含量以及较高比例的低沸点化合物,导致其具有较低的分子量。
在一个实施方案中,本文提供了用于共同生产生物气和生物原油的方法,包括:
a)将废水污泥与废弃木质纤维素生物质混合以形成总固体含量为约5至约25wt%的混合物;
b)使混合物在保持温度为约200℃至约350℃,约50巴至约150巴的压力以及存在催化剂的的反应器中进行水热液化,以得到反应产物;
c)从反应器的反应产物中除去并收集固体生物炭,从反应器的反应产物中除去并收集生物油,并且从反应器的反应产物中除去并收集水性产物;和
d)厌氧消化水性产物以产生并收集由厌氧消化的水性产物所产生的生物气。
在该方法的一个实施方案中,废水污泥和废弃生物质的混合物可具有约8wt%至约20wt%的固体含量。
在该方法的一个实施方案中,废水污泥和废弃生物质的混合物的固体含量可为约10wt%。
在该方法的一个实施方案中,温度可以保持在约280℃至约330℃的范围内。
在该方法的一个实施方案中,压力可以保持在约100巴至约150巴的范围内。
在该方法的一个实施方案中,催化剂可以是koh、k2co3、naoh、na2co3、硬硼钙石、feso4、ca(oh)2、水滑石(ht)和mgo中的任一种或它们的组合。
本方法可以间歇模式或连续操作模式进行。
通过参考以下详细描述和附图,可以实现对本公开的功能和有利方面的进一步理解。
附图的简要说明
从以下结合附图的详细描述中将更全面地理解本文公开的方法的实施方案,附图是本申请的一部分,并且其中:
图1显示了本文公开的方法的框图。
图2显示了100ml和500ml间歇反应器的示意图表和照片。
图3显示了用于实施本发明方法的连续流动反应器的非限制性实施方案的示意图。
图4显示了由was和液体纤维素生物质的共液化产生的生物油的ft-ir光谱。
图5a显示了本实施例中使用的原料的热重分析(tga)曲线。
图5b显示了本实施例中使用的原料的差热分析(dta)曲线。
图6显示了使用本发明方法生产的生物油的tga曲线。
图7a显示了来自桦木锯屑(bs)和废活性污泥的混合物的生物油的tga曲线。
图7b显示了来自玉米秆(cs)和废活性污泥(was)的混合物的生物油的tga曲线。
图7c显示了来自废报纸(np)和废活性污泥的混合物的生物油的tga曲线。
图8显示了水溶性产物(wsp)的生物化学甲烷势(bmp)结果。
具体实施方式
将参考下面讨论的细节描述本公开的各种实施方案和方面。以下描述和附图是对本公开的说明,而不应被解释为限制本公开。描述了许多具体细节以提供对本公开的各种实施方案的透彻理解。然而,在某些情况下,没有描述众所周知的或传统的细节以便提供对本公开的实施方案的简明讨论。
如本文所用,术语“包括(comprise)”和“包含(comprising)”应被解释为包含性的和开放性的,而不是排他性的。具体地,当在说明书和权利要求书中使用时,术语“包括(comprise)”和“包含(comprising)”及其变体“表示包括指定的特征、步骤或组件。这些术语不应被解释为排除其他特征、步骤或组件的存在。
如本文所用,术语“示例性”表示“用作实例、例子或说明”,并且不应被解释为比本文公开的其他配置更优选或更具优势。
如本文所用,术语“约”和“近似”旨在涵盖可能存在于值的范围的上限和下限中的变化,例如性质、参数和尺寸的变化。在一个非限制性实例中,术语“约”和“大约”表示加或减10%或更少。
如本文所用,短语“生物气”主要是指甲烷(ch4)、二氧化碳((co2)中的一种或两种,然而应该理解,生物气是主要为甲烷和二氧化碳的混合物,其中含有痕量的其他气体,例如氮气、氢气、硫化氢和氧气。
如本文所用,短语“生物原油”或简称“生物原油”或“生物油”是来自生物质(或生物原料)的水热液化的油性产物。它是在高压和高温下并且在缺少氧气的情况下停留时间为数分钟至数小时的通过水热处理生物质所回收的高粘度液体(或固体状)冷凝物。
如本文所用,短语“废水污泥”通常是指在处理流出物(例如一级或二级处理)期间在不同步骤中产生的来自废水流(城市或工业)的固体残余物。废水污泥还可以包括其他含有高水分的生物质,例如但不限于湿的收获的海藻类生物质。
如本文所用,短语“废物生物质”是指各种类型的木质纤维素生物质,例如林业残余物、城市固体废物、木材废物和农业残余物或废物。
以下提供实施例中常用的材料和方法。
材料
桦木和橡胶木屑由安大略省伦敦的一家当地木材厂供应。玉米秆来自当地农场。将原料研磨成平均尺寸小于20目的颗粒。在当地收集的旧报纸被用作废报纸。将收集的废报纸在水中浸泡24小时(h),然后用家用搅切机压碎成纸浆。然后将纸浆在105℃下干燥12小时,用wiley磨机研磨成<20目的颗粒,并储存以备将来使用。废活性污泥(was)从安大略省伦敦的阿德莱德污染控制厂收集。was样品取自旋转鼓式增稠器,并在实验前在4℃下储存。实验中使用的催化剂是购自sigma-aldrich的氢氧化钾(koh),并按原样使用。
虽然催化剂可以是koh、k2co3、naoh、na2co3、硬硼钙石、feso4、ca(oh)2、水滑石(ht)和mgo,但最经济可行的催化剂可以是koh、k2co3、naoh和na2co3中的任何一种或它们的组合。
a.c.s.试剂级丙酮,用作产物分离的反应器冲洗/洗涤溶剂,购自caledonlaboratorychemicals并按原样使用。
实验设置
实验在间歇和连续流动反应器中进行。
实施例1至实施例4在间歇反应器中进行。间歇反应器是100ml和500ml搅拌反应器50(parr4590和4570微型台式反应器),配备有混合器(m)、加热器、热电偶、温度控制器和压力计(pg),参见图2。对于每个实验,将适量的木质纤维素生物质加入到40g的was中(在干燥和无灰基础上,使固体浓度为5-15wt%)并将该混合物与koh(总固体为5wt%)一起加入到反应器作为均相催化剂,基于作者进行的先前的催化剂筛选研究来进行选择[10]。由于was含有约96wt%的水,没有外部水加入到反应混合物中作为溶剂。然后,密封反应器50,并通过用氮气吹扫至少五次来除去内部的残余空气。然后,使用氮气将反应器50加压至2mpa,然后在搅拌下加热至所需温度(200℃-350℃)。一旦反应器50达到反应温度,就在该温度下保持所需的保留时间(10-60分钟)。此后,通过在水/冰浴中淬灭反应器50来终止反应。采用相同的程序用于在最佳操作条件下的实验。
实施例5在连续流动反应器中进行。为本发明的方法设计和构建连续反应器设置。图3显示了用于水热液化的连续流动反应器系统10的示意图。该系统的主要部件包括5/8英寸的ss316l管式反应器12、两个活塞进料器14、进料罐16、hplc泵18、预加热器20和加热器22、冷却器24、两个气液分离容器26和背压调节器30。向进料罐16中加入制备的浆料,并使用压缩空气将其填充到活塞进料器14中。然后,通过使用由hplc泵18供应的高压水驱动的活塞进料器14泵送,将进料注入反应器10中。然后,系统由来自罐36的氮气加压,并由加热器20和22进行加热,以达到所需温度。通过背压调节器30将压力调节到所需压力。在通过反应器10后,进料将在冷却器中冷却,通过分离容器26并闪蒸。气体将从顶部离开分离容器26,并且将从容器26的底部收集固体/液体产物。给予系统足够的时间(约2小时)以达到所需的温度和稳态操作。来自第一活塞进料器14的进料用于过程稳定化,并且流出物被收集于第一分离容器26中。一旦操作条件稳定,就切换活塞进料器14以从另一个活塞进料器14供给反应器10。在第二分离容器26中收集htl产物。
对于连续流动反应器中的实验,为了防止生物油产品粘附到反应器壁上并堵塞反应器,使用乙醇作为水中的共溶剂用于通过溶剂原位提取生物油或反应中间体。通过混合1000g的污泥、木屑(木屑与污泥的质量比为0.15∶1(w/w)),1.4g的koh(基于干燥、无灰的底物的5wt%)以及相对于包括乙醇和was的反应混合物的总重量的30wt%的乙醇来制备原料。为了促进was和具有高固体浓度的木屑的共同进料的泵送,将羧甲基纤维素钠(cmc)以总反应混合物的3wt%的量添加到原料浆液中以获得均匀的悬浮液。进料具有12wt%的无灰固体浓度。
产品分离程序
在将反应器50冷却至室温后,将反应器50中的气体收集到1.0l气囊中用于gc-tcd(agilentmicro-gc3000)分析(120ml空气作为内标注入气囊中)。然后,打开反应器50,并从反应器50中取出固/液产物并转移到离心管40中。将它们以4500rpm离心10分钟,并在真空下通过预先称重的0.45μ.玻璃纤维滤纸过滤。收集滤液作为水溶性产物(wsp)。然后,用来自清洁溶剂罐42的试剂级丙酮冲洗反应器50,以通过用刮刀刮擦完全除去任何剩余的材料,包括生物原油和粘附在反应器内壁上的残余炭。收集浆液和冲洗丙酮并在真空下通过0.45μm玻璃纤维过滤,在其上保留水不溶性固体。用丙酮冲洗总固体残余物直至产生的滤液变为无色。然后,将总固体残余物在105℃下烘箱干燥过夜至恒重,以确定固体残余物(sr)和生物质转化的产率,同时在减压下蒸发滤液以于50℃在旋转蒸发器中除去丙酮,并且将留下的深色产物称重并指定为生物原油。
间歇反应
对于间歇反应,然后基于干燥、无灰(daf)的初始生物质计算产物的产率,如下:
wsp的产率(wt%)=100-(生物油的产率+气体的产率+sr的产率)(4)
连续流动反应
对于连续流动反应,产率计算如下:
然后基于干燥、无灰(daf)的初始生物质计算htl产物的产率,如下:
wsp的产率(wt%)=100-(生物油的产率+气体的产率+sr的产率)(8)
其中mb是生物油的质量,
实验设计
使用响应面方法(rsm)进行实验设计。rsm是使用来自实验的定量数据来建模和分析问题的统计方法,以通过回归来确定模型方程。该方法优化了对工艺参数变化的响应[11],[12]。中心复合设计(ccd)是最受欢迎的rsm设计之一,可用于构建响应变量的二阶(二次)和三阶(三次)模型。二次方程的一般形式可表示如下[12]:
其中y是响应,b0是常系数,bi、bii和bij是线性、相互作用和二次系数,且xi、xj分别是独立变量的编码值[12]。在目前的工作中,应用具有三个变量的标准ccd设计,以研究三个独立变量(温度、时间和固体浓度)对生物油产率的影响。该设计包含8个立方点、6个轴向点和1个中心点,其中中心点重复6次。因此,总共进行了20次实验。选择中心点重复作为精确度的度量。变量水平在温度为200℃-350℃,反应时间为10-60分钟,固体浓度为5wt%-15wt%的范围内。因子和水平示于表1中。对于统计计算,变量xi根据以下关系编码为xi:
其中hi是特定变量的未编码的高水平,lo是未编码的低水平。
使用designexpert(版本7)和minitab(版本17)软件分析设计矩阵,并且进行优化以最大化生物油产率。还测定了油的分子量(mw)和其他产物如固体残余物、wsp和气体的产率,以探索不同操作条件对产物产率的影响。
产品分析
原料和产物的元素分析在flashea1112分析仪上进行,使用2,5-双(5-叔丁基-苯并噁唑-2-基)噻吩(bbot)作为校准标准。通过差值估计氧气的组成。基于dulong公式
通过perkinelmerpyris1tga在氮气和空气气氛中测定热重(tga)分析、挥发性物质(vm)和固定碳(fc)含量。将样品从40℃加热至900℃,加热速率为10℃min-1,然后在空气中在900℃下燃烧20分钟。气体流速为20mlmin-1。使用总有机碳(toc)分析仪(shimadzutoc-asi)测量水溶性产物中的总有机碳含量。
根据标准方法进行水溶性产物的总固体(ts)、挥发性固体(vs)和总需氧量(tcod)[14]。水分含量和灰分含量分别根据astme1756-08(于105℃干燥样品至少12小时)和astme1755-1(于575℃加热样品3小时)测定。
通过使用sianalytics电位滴定仪的ph探针测定was样品的ph。傅立叶变换红外光谱仪(ft-ir)分析在perkinelmerft-ir光谱仪上进行,并在4000-550em-1的范围内记录光谱。
使用电感耦合等离子体(icp-aes)测定灰的化学组成。固体样品用硝酸和硫酸在90℃下进行酸性消化1小时。然后将它们冷却至环境温度,然后在icp分析之前过滤和稀释。在icp测试期间,将样品加热至6000-8000k,以使金属化合物蒸发并电离以进行定量:na、k、mg、ca、mn、fe、zn、al和si。通过原子发射光谱法检测和分析离子。
生化甲烷生产测试
使用amptsii(bioprocesscontrol,sweden)测量生物化学甲烷势(bmp)。将间歇厌氧反应器用从当地市政厌氧消化池收集的消化液(vs约1.1%)接种,并以质量vs为基础按底物与接种物比为约1∶3接种底物。wsp(底物)和厌氧种子的体积分别为约50和450ml。单独的种子用于空白bmp测试。bmp测试进行约30天,然后停止。
原料表征
原料样品的生理化学特征在表2中给出。对原料的近似分析表明,与报纸(76.1%)、玉米秆(74.1%)和was(62.2%)相比,桦木锯屑基于干重(83.5%)的总挥发性物质含量最高。木质纤维素生物质的有机物主要由木质素、纤维素和半纤维素组成,而废水污泥主要是蛋白质、脂类和碳水化合物。与挥发性物质相比,废活性污泥的灰分含量高达23.67%,相比之下,木屑灰分(0.23%)的量可忽略,玉米秆为10.7%且报纸为9.2%。通过icp-aes分析灰分中的无机矿物质,结果如表3所示。分析表明,灰分的主要成分是木质纤维素生物质的钙、钾和镁,以及废活性污泥的铁和钙。
原料的元素分析表明,与木质纤维素生物质相比,污泥中的氮浓度更高,这很可能是由于蛋白质的存在。蛋白质也含有硫,并且还有一些含硫氨基酸,例如蛋氨酸和半胱氨酸[15]。因此,在液化产物中可以预期含氮和硫化合物(由于蛋白质的热降解而形成)。对于所有样品,h/c和o/c的摩尔比为1.57-1.65和0.48-0.75,分别具有14.6-16.9mj/kg的低的高热值(hhv)。
实施例1
使用was和锯屑(bs-was)的混合物进行实验设计研究。使用标准ccd设计进行优化以使生物油产率最大化。由于从废物利用的观点来看,较高浓度的原料更有益,因此对优化过程施加x3>10wt%的约束。使用如表4中所示的实验数据(两次重复实验)验证推荐的最佳操作条件。实验和预测值非常一致,表明模型具有良好的可预测性。
实施例2
将玉米秆和废活性污泥(cs-was)的混合物在实施例1中获得的最佳操作条件下进行液化(310℃的温度,10分钟的反应时间和10wt%的底物浓度,在作为均相催化剂的koh存在的情况下)。表5中给出了产物的产率、转化百分比和生物原油的分子量。
实施例3
将报纸和废活性污泥(np-was)的混合物在实施例1中获得的最佳操作条件下进行液化(310℃的温度,10分钟的反应时间和10wt%的底物浓度,在作为均相催化剂的koh存在的情况下)。表6中给出了产物的产率、转化百分比和生物原油的分子量。
将实施例1至实施例3的产物产率进行比较,bs-was和cs-was产生最高量的生物油,且cs-was导致最低量的固体残余物,因此转化率最高。作为一般趋势,htl条件下不同生物质成分的转化率的次序为脂质>蛋白质>碳水化合物[16]。碳水化合物的低转化率主要是由于较高的半纤维素和木质素含量。np-was的较低生物油产率是由较高碳水化合物含量来解释,其具有较低的转化效率。通常,与玉米秆和锯屑相比,报纸具有更高百分比的纤维素和木质素[17]-[22]。这也通过在后面章节中所讨论的原料的tga分析得到证实。使用cs-was的实验的最高转化率也可能是由于与其他两种木质纤维素生物质相比,玉米秆的木质素含量最低(7.3-16%)[17],[18]。先前的研究表明,木质素的水热加工增加了固体产量,因为木质素解聚后随后进行再聚合或自缩合[23],[24]。
cs-was的较高转化率也可归因于玉米秆灰中存在无机材料,其可起到催化剂的作用以提高转化率并导致固体残余物产率降低。矿物盐可以加速中间产物的二次解聚并防止固体残余物的形成。如先前在表2中所示,与锯屑相比,玉米秆和报纸的灰分含量高得多。对灰分的分析表明,与其他原料相比,玉米秆的钾含量最高。据报道,钾的存在对于抑制水热液化过程中的固体产率是有效的。碳酸钾可导致固体残留物减少,而氢氧化钾可促进水煤气变换反应[23],[25]。钠盐还可以提高生物原油产率并抑制炭的形成,然而它们的活性低于钾盐[23]。诸如fe或ni的微量元素也可能导致使用玉米秆和was的混合物的实验的固体产率降低。
实施例4
将来自桦木锯屑(bs)或was液化的油产率列于表7中以进行比较。将来自bs和was的油产率与bs-was的油产率(实施例1)进行比较,表明了向was添加锯屑对油产率没有协同作用。然而,关键的发现是,如果将额外的木质纤维素生物质(例如,锯屑、玉米秆、msw)作为共同进料添加到废水污泥中,则生产的生物原油将具有比使用单独的木质纤维素生物质生产的生物油更低的分子量和更高的能量含量,这表明了木质纤维素生物质和废水污泥的共液化的协同作用和有利效果。
实施例5
在实施例1中获得的最佳操作条件下,将橡胶木屑和废活性污泥的混合物进行液化(310℃的温度,10分钟的反应时间,在作为均相催化剂的koh存在的情况下)。将固体浓度增加至12%以检查浆料在较高浓度下的流动性。表8中给出了产物的产率、转化百分比和生物原油的分子量。
实施例1至实施例4中所生产的生物油的特征
分子量
通过gpc测量生物原油的分子量,对于来自混合物的生物油,其分子量为448-562g/mole,且对于cs-was样品,具有448g/mole的最低的分子量。在作者先前的htl实验中[10],在几乎相同的操作条件下使用锯屑作为原料(koh作为催化剂,300℃的温度,10wt%的固体浓度和30分钟的反应时间)。发现该生物油的分子量为856g/mol。与此相比,was和木质纤维素生物质的混合物导致油的分子量低得多,因此粘度较低。与一些研究人员先前报道的来自污泥或锯屑的生物油相比,cs-was油的分子量也较低。例如,vardon等人,研究了包括螺旋藻、猪粪和消化厌氧污泥的三种废弃物原料于300℃、30分钟的反应时间和10-12mpa压力的情况下的水热液化。发现ts=26%的来自污泥的生物油具有最高的分子量(3470g/mol)[13]。表9列出了文献中的生物油分子量的一些结果。似乎较高的固体浓度可能由于更多的聚合反应而产生更高分子量的生物油。虽然藻类的木质素含量远低于木屑,但htl实验中使用的高固体浓度可能导致产生更高分子量的生物油。cs-was生物油的较低分子量表明由于was和木质纤维素生物质之间的化学反应而产生较轻的化合物。更详细的生物油表征,例如化学成分、官能团、热稳定性等,将在下一节中讨论。
较高的热值
表10列出了用不同原料生产的生物原油和固体残余物的元素分析及其较高的热值(hhv)。列出了was或锯屑的htl结果用于比较。来自混合物的生物原油的碳含量(69.1-72.4%)远高于原始生物质材料(38-47.6%)的碳含量。此外,油的氧含量为16.3-22.1%,远低于原料中的24.4-45.9%,导致油的增加的较高的热值。生物原油产品的hhv为26.4-32.4mj/kg,相比之下,粗原料的hhv仅为14.6-16.9mj/kg。原料的类型对生物原油的h/c和o/c比具有很大影响。来自was和木质纤维素生物质的混合物的生物油的o/c摩尔比介于来自was的生物油和来自bs的生物油的o/c比之间,与来自bs的生物油的o/c和h/c比相比较,bs-was和np-was具有低得多的o/c比和较高的h/c比。bs-was和np-was显示出相似的组成,因此具有相似的较高的热值,然而来自cs-was的生物油具有较高的氧和较低的氢含量,导致较低的hhv。通常,与进料的初始h/c比(1.57-1.65)相比,油的h/c摩尔比(0.85-1.26)降低。较低的h/c摩尔比表示脱氢反应,例如醇和胺的脱氢以及醛和酮的产生。
因此,在生物油中预期由于醛的脱氢而存在这些化合物以及羧酸衍生物。较低的h/c摩尔比也表明了油中的高度不饱和结构。所有生产的油的o/c比(0.17-0.24)远低于生物质进料(表2中的0.48-0.75),表明了在水热液化期间反应中间体发生脱氧反应(脱水或脱羧反应),导致气态产物中产生wsp和co2[28]。在实验中大量的水(50-60wt%)形成为wsp。而且,根据micro-gc分析,气体产物的主要成分是co2。这表明在液化过程中主要以co2和wsp的形式去除生物质中的氧。
固体残余物的元素组成表明,炭的氢含量为2.2%至3.9%。碳主要以焦炭形式的炭存在,含量为25.3%至50.7%。炭的h/c摩尔比为0.92-1.1,表明主要存在芳族化合物。此外,氧主要存在于灰成分中,与金属氧化物形式的金属元素结合,金属氧化物在整个过程中无活性。
将这些固体与来自锯屑实验的固体残余物比较表明,尽管存在较高的h/c值和显著较低的氧含量,但由于来自共进料的固体残留物中存在高灰分,因此热值较低。固体残留物可用作其他工厂操作的能源。然而,灰分应该从固体中除去,然后它们才能用作固体燃料产生热量,因为灰分在焚烧后仍然是残余物,且高灰分含量可引起严重的腐蚀问题。
锯屑所生产的生物原油与含有从共同进料(玉米秆和was实验除外)获得的油的生物原油的元素组成表明了油的碳和氢含量的改善,以及当使用混合物时,氧含量下降随后导致油的热值显著增加。这可能是由于was的存在引起的协同效应,因为与来自锯屑的生物油相比,来自废活性污泥的生物油具有非常高的c碳和氢含量以及显著低的氧百分比。来自锯屑的生物油的h/c摩尔比为1.1,表明存在芳族化合物,因此该油的粘度更高。另一个重要的区别是,用共同进料生产的油中氮的浓度较高且存在硫。这是由于与根据表2中的其他类型的原料相比,was中的高含量的硫和氮,其也存在于来自was的油中。与锯屑产生的油的氮含量(0.1%)以及较高的硫含量相比,was的高蛋白质含量被带到生物原油中并导致高氮含量(3.1-3.6%)。
然而,与硫含量范围为0.1%至3%的许多石油原油相比,硫含量仍然相对较低[29]。与具有0.05-1.5%的氧和0.01-0.7%的氮的石油相比,油的氮和氧含量仍然太高。杂原子含量是区分生物原油和石油的主要因素[13],[16]。为了改善这些生物油的质量,需要进一步的升级过程以进一步降低氧含量和酸含量。
为了更好地了解原料,产品中的碳分布,该过程的材料平衡通过碳平衡进行,并且列于表11中。通过元素分析确定生物原油和固体残余物的碳组成,并分别通过总有机碳(toc)和微量gc分析获得wsp和气体产物的碳含量。基于产品中碳的质量百分比与干燥原料中的碳质量的关系计算碳回收率。如表11所示,总碳回收率在89-99%的合理范围内。
如上所述,原料中碳的最大部分被转移到生物原油中。较小的部分最终形成水溶性产物,只有非常小的部分转移到固体中。使用bs-was获得最佳的碳回收率(99.66%)。在一些试验中较差的质量平衡可能是由于在收集生物原油产品的蒸发过程中损失了一些低沸点和低分子量有机物[30],[31]。
生物原油官能团
进行4000-550cm-1范围内的生物原油的ft-ir分析以鉴定官能团,结果显示在图4中。无论生物质的类型如何,所有生物油都显示出相似的官能团。差异仅在于峰的强度。在3350cm-1处的宽吸收是o-h伸缩的典型吸收,表明生物原油中存在醇、酚、羧酸和水残余物。它也归因于蛋白质基团的n-h伸缩。3000和2840cm-1之间的带表示c-h伸缩振动,表明存在烷基c-h。根据图4,用共进料生产的油的这些峰的强度强于来自锯屑的油,表明这些油中存在更多的烷基。然而,与来自was的油相比,它们较弱,表明该油具有更大量的烷基。1700cm-1处的吸光度表示羰基的c=o伸缩振动,表明油中存在酮、醛和羧酸。在1611cm-1、1516cm-1和1456cm-1处的峰表示芳环及其衍生物。这些峰的强度,尤其是在1611cm-1和1516cm-1处的峰强度在来自锯屑的油中更强,表明该油含有更多这些化合物。1280cm-1至1000cm-1之间的带可归因于c-o振动,表明生物油中可能存在酸、酚或醇。在1370cm-1和1456cm-1处的两个吸收分别归因于甲基(-ch3)和亚甲基(-ch2)的弯曲峰。
生物原油的化学组成
通过gc-ms表征油产物以鉴定它们的化学组成。应该注意的是,由于生物油的高分子量和沸点分布以及仪器的温度极限(检测到最高沸点300℃),通过gc-ms仅可鉴定由htl形成的一小部分产物。此外,一些低沸点化合物可能已被溶剂峰掩盖或在蒸发用于回收生物原油的丙酮时损失[13]。
含氮化合物、脂肪酸和酚类构成了bs-was和np-was生物油的主要部分,而cs-was中生物油的最大部分是酯类,其次是脂肪酸和含氮化合物。在油中鉴定了其他组分,例如烷烃、醇、胺、酰胺、苯化合物、羧酸和酮。在bs-was油样品中发现了最高比例的酚类化合物,其次是np-was和cs-was样品。酚类化合物如2-甲氧基-苯酚和4-乙基-2-甲氧基-苯酚主要来源于通过裂解木质素中的芳基醚键而降解的木质素组分。它们也可以衍生自碳水化合物和蛋白质部分[16]。与锯屑和报纸相比,玉米秆通常具有较低的木质素含量。因此,来自cs-was的油在其他油之中具有最低量的酚类化合物。was的蛋白质含量导致生产出具有高百分比的含氮化合物的生物原油。
在油中存在这些化合物如1-十二胺、2-甲基丙酰胺和1-乙酰基-4-[1-哌啶基]-2-丁炔酮表明,蛋白质因水热液化通过氨基酸的脱羧和重排而降解。含氮有机化合物可能通过美拉德反应与糖反应形成吡啶[32]。生物油样品中吡啶的存在证实了该反应的发生。酯类是从cs-was获得的油的主要成分。源自纤维素分解的呋喃衍生物的分解可能有助于酯的形成。所有的油都含有相当大比例的脂肪酸,这些脂肪酸是由was中的脂质分解产生的。
将这些油的成分与来自单独的木屑和单独的was的油进行比较表明,使用was和木质纤维素生物质混合物生产的油含有的酚类化合物比来自锯屑的油少得多,与来自单独的锯屑和单独的was的油相比,含有相当更高的量的酯类,并且来自锯屑的油相比,脂肪酸、含氮化合物和饱和化合物的百分比高得多。油中较低的酚类成分可归因于污水污泥中较低的木质素含量。用was和木质纤维素生物质混合物所生产的油中苯和苯衍生物的含量非常低,远低于酚类化合物;然而,它们仍然高于来自锯屑的油,这表明在与was和木质纤维素生物质的混合物的反应中苯环上的酚的-oh更容易被除去。与来自共同进料的油相比,用锯屑生产的油中芳烃(包括苯衍生物、苯酚和苯甲醛)的总百分比要高得多,这也在ft-ir分析中得到证实。
热重分析
通过tga测量原料和油的热稳定性。在分析之前,将样品在60℃下烘箱干燥1小时。然后,在热重分析仪上在n2气氛下将它们从40℃加热至900℃,并连续记录样品的重量损失(tg)和重量损失率(dtg)。然后,将气体转换为空气,并将样品在空气中在900℃下燃烧20分钟,以确定其固定碳(fc)和灰分含量。
原料的热重分析
不同原料的tg和dtg曲线如图5所示。所有三种木质纤维素生物质原料具有相似的分解曲线(tg),由于其较高的挥发性物质含量,锯屑的重量损失更多。然而,它们明显不同于was的tg图。污泥曲线和木质纤维素生物质曲线之间的差异是由于不同的有机和无机物质特征。通常已知生物质材料主要由蛋白质、碳水化合物、木质素和脂质组成。如已提及的,污泥主要由蛋白质、脂类和碳水化合物组成,而木质纤维素生物质主要包含碳水化合物和木质素。锯屑、报纸和玉米秆的结构在约280-300℃开始分解,可能与其半纤维素含量有关,然后在300-400℃开始更快地降解,最可能与其纤维素含量有关。然而,污泥的分解开始于约200℃,其比木质纤维素生物质低80-100℃,较浅的陡峭表明污泥的挥发性物质含量较低。污泥的分解曲线分两个阶段进行:第一阶段在200-370℃是由于细胞中存在可生物降解的物质和有机聚合物,第二阶段在370-500℃是由于不可生物降解的材料,如纤维素和类似材料。
不同原料类型的热分解之间的差异也可以从dtg曲线的形状确定。原料样品的曲线在约100℃的温度显示出轻微的重量损失峰值,这可归因于剩余水分的脱水和样品中轻质挥发性化合物的释放。木质纤维素原料的最大降解速率发生在330-370℃,表明纤维素的分解在样品中占主导地位。峰的相对强度可以与原料中存在的组分的总量相关。在木质纤维素生物质样品中,发现锯屑具有最高的纤维素含量。在450-500℃和620-730℃之间的较小dtg峰中有一些木质素的迹象,其更稳定,并且具有280-500℃和175-800℃的更宽的降解温度[33]。根据dtg图表,与玉米秆或锯屑相比,报纸的木质素含量要高得多。was的降解发生在两个阶段:蛋白质和半纤维素在第一阶段(200-370℃)的热分解和蛋白质和纤维素在第二阶段(370-500℃)的热分解。峰的强度表明,与木质纤维素生物质相比,was具有低得多的纤维素和半纤维素含量。
生物油的热重分析
油的tg和dtg图显示在图6和图7中。从tg/dtg曲线获得的一些关键参数,即初始分解、最终分解和峰值温度以及挥发性物质(vm)和固定碳(fc)的含量示于表12中。根据tg图,在由was和木质纤维素生物质的混合物所生产的生物油之间的热稳定性没有显著差异。然而,与来自锯屑的生物油的曲线(212℃)相比,这些生物油的分解曲线转移到较低温度(161-168℃)。它甚至低于来自was的生物油的分解温度(208℃)。该结果表明它们具有较低的热稳定性并且需要较低的活化能来分解这些油。与从单独的锯屑生产的油的59.3%的vm和40.7%的fc相比,它们还具有较高的挥发性物质含量(71-77%)和较低的固定碳含量(22-28%)。由于来自was的油也显示出非常高的vm含量(86.9%),因此当was和木质纤维素生物质用作共进料时,来自混合物的生物油的增加的vm含量可能是由于协同效应。
dtg曲线根据重量损失率分为数个阶段,即阶段“a”是表面水分的脱水和轻组分的蒸发,阶段“b”是低分子量材料的脱挥发分和蒸发,阶段“c”是聚合和脱水,最后阶段“d”是炭分解阶段。阶段和温度范围显示在图7a、图7b和图7c中。由于油在烘箱中预干燥,因此阶段a显示出小峰。对于bs-was和cs-was,低分子量材料的挥发范围更宽从约100℃开始到250-300℃。因此,与np-was相比,阶段b和c变得更加明显。来自np-was的油具有较低量的轻组分。在阶段c中,生物原油聚合成缩合材料如树脂以及加热时发生重馏分的脱水和缩合。与np-was相比,bs-was和cs-was显示出更高的峰,表明更多的重馏分被分解成这两种油。最后的分解阶段更宽泛,并且伴随着np-was的非常大的峰值,表明在前一阶段加热该生物油期间产生更多的炭。
tga数据也可用于估算重油的沸程[34]。使用热重分析数据确定生物原油的沸点分布,并且列于表13中。在110℃之前样品的重量损失是水分的指标,并且对于所有油都小于2wt%,这表明干燥过程有效地去除了水。根据表13,与单独的锯屑相比,was和木质纤维素生物质的混合物具有较低沸点的组分的百分比增加。在was存在下生产的约30-37wt%的生物油具有低于300℃的沸点,而用单独的锯屑生产的油中仅为19wt%。这意味着添加was已将分子分布转变为更易挥发的化合物。
实施例5
通过bmp分析,将水溶性产物(wsp)用于甲烷生产。bmp是确定生物质用于厌氧消化的潜力的重要且有价值的测定法。首先分析wsp样品的toc、cod、vs和ts。wsp的固体浓度可以忽略不计。样品的总固体(ts)和挥发性固体(vs)分别为1.52%和0.84%,且toc和cod分别为16.33g/l和41.86g/l,使得cod/toc比为2.5。该比表示由于htl处理的碳化合物的还原程度。图8显示了每添加挥发性固体(vs)(g)的来自wsp的累积甲烷产量。bmp结果显示快速的初始甲烷产量(没有滞后期),在31天后添加的每克(g)vs的约800ml的峰值。由于50ml的wsp用于bmp测试,因此产生的气体体积为每0.816g的总有机碳(toc)或2.09g的cod。基于cod测量的样品的可降解性为46%。
水溶性产物是来自水热液化过程中的副产物的最大的部分。直接从共液化过程中使用该副产物而无需任何进一步处理以产生生物气是本研究中产生的新方法。结果表明,该副产物可以产生大量的生物气,使生物气和生物油的共生产成为可能。产生的生物气可用于产生电和热,其中产生的能量可以再投回到过程中。
结论
本文已经公开了基于水热液化(htl)处理的方法,用于共同处理高含水量废水污泥和其他木质纤维素生物质,以共同生产生物气和生物原油。已经基于最大生物原油产量的响应面方法优化操作条件,包括反应温度、反应时间和固体浓度。将三种类型的木质纤维素废弃生物质(桦木锯屑(bs)、废报纸(np)和玉米秆(cs))与废活性污泥(was)混合,并在优化的操作条件下转化为生物原油。这些废生物质材料仅是示例性的,本发明的方法不限于它们。
废活性污泥和其他废生物质的共转化是将两种类型的废料同时转化为具有附加值的产品的有益方法,其优点是与木质纤维素生物质相比生产了更高质量的生物原油。当污泥与木质纤维素生物质混合时,与来自锯屑的生物油(856g/mol)相比,所生产的生物油的分子量显著降低(448-562g/mol),表明was和木质纤维素的协同作用,导致生物油中存在较轻组分。
根据vankrevelen图,来自共同进料的生物油呈现较低的h/c和o/c比,表明脱氢和脱氧反应的发生,这导致更高质量的生物原油。根据gc-ms结果,与由锯屑生产的油相比,由共同进料生产的油具有更少的酚类化合物,相当高的酯、脂肪酸和含氮化合物的量。
与由锯屑生产的生物油相比,用共同进料生产的生物油具有更高的挥发性物质含量和更低的固定碳。它们还表现出较低的热稳定性以及因此的较低的分解活化能。与用锯屑生产的油中只有19wt%相比,这些油的沸点分析表明,存在30-37wt%的低分子量化合物(<300℃),这导致这些油的分子量显著降低。
该过程的两种副产物可用于产生热量和电力。固体残余物或生物炭可用作发热的固体燃料。wsp可用于生产生物气厌氧消化。bmp测试显示,每0.816g的总有机碳(toc)或2.09g的水溶性产物的化学需氧量(cod),在30天内累积产生800ml的生物甲烷。
由于数个原因,本方法与先前的废水htl方法显著不同。首先,本方法旨在共同生产生物气和生物原油。使用其他废料如锯屑、玉米秆、msw等共处理废水污泥(含水量超过90%)以将底物浓度调节至最佳值,从而提高该方法的经济性。而且,这使得该方法能够同时处理两种类型的废物生物质。来自该过程的生物原油的产率显著高于stors过程的产率。
此外,本文公开的共同生产方法使用htl(水溶性产物)的副产物用于生物气生产,其不同于cambi或biothelys方法,其中污泥用于厌氧消化和生物气生产。与cambi或biothelys方法相比,本发明的水溶性产品加工方法不需要干燥或脱水,通过消除昂贵的干燥过程提供成本优势。
此外,用于液体燃料的htl生物油的质量高于快速热解油,具有较低的水分和较高的能量含量。
最后,可以容易地使用过程副产物,例如固体生物炭可以是其他工厂操作的能源或可出售,并且水溶性流可以用于通过厌氧消化生产生物气或再循环回到水处理过程中。
表格
表1
实验变量和水平
表2
原料的特性
a-在干燥的基础上
b-通过tga于800℃在氮气和空气气氛中测定
c-在实验之前,将原料在105℃的烘箱中干燥24小时
d-按差值计算(100%-c%-h%-n%-s%-灰分%)
e-通过dulong公式计算的较高的热值(hhv),即,hhv(mj/kg)=0.3383c+1.422(h-o/8)
表3
通过icp-aes检测原料灰分中主要无机元素的浓度
nd:未检测的
表4
最佳操作条件,预测的和实验的油和固体的产率
表5
来自cs-was的htl的生物原油的产物分布和分子量,在存在koh的情况下于310℃,10分钟以及10wt%固体浓度
表6
来自np-was的htl的生物原油的产物分布和分子量,在存在koh的情况下于310℃,10分钟以及10wt%固体浓度
表7
来自bs和was的htl的生物原油的产物分布和分子量,在存在koh的情况下于310℃,10分钟以及10wt%固体浓度
1结果获得自作者在几乎相同的操作条件下(300℃,30分钟和10wt%固体浓度)的以前的研究
表8
表9
由污泥或木质纤维素生物质所产生的生物油的分子量
表11
于310℃液化10分钟的产品中的碳回收率。
表12
分解开始/峰值/终止温度,挥发性物质和生物原油的固定碳
表13
生物原油的估算沸点分布(%)
参考文献
[1]p.azadi,e.afif,h.foroughi,t.dai,f.azadi,andr.farnood,“catalyticreformingofactivatedsludgemodelcompoundsinsupercriticalwaterusingnickelandrutheniumcatalysts,”appl.catal.benviron.,vol.134-135,pp.265-273,may2013.
[2]e.neyens,j.baeyens,andc.creemers,“alkalinethermalsludgehydrolysis.,”j.hazard.mater.,vol.97,no.1-3,pp.295-314,2003.
[3]e.neyens,j.baeyens,r.dewil,andb.deheyder,“advancedsludgetreatmentaffectsextracellularpolymericsubstancestoimproveactivatedsludgedewatering,”j.hazard.mater.,vol.106,no.2-3,pp.83-92,2004.
[4]c.he,a.giannis,andj.-y.wang,“conversionofsewagesludgetoclcansolidfuelusinghydrothermalcarbonization:hydrocharfuelcharacteristicsandcombustionbehavior,”appl.energy,vol.111,pp.257-266,nov.2013.
[5]v.k.tyagiands.-l.lo,“sludge:awasteorrenewablesourceforenergyandresourcesrecovery,”renew.sustain.energyrev.,vol.25,no.71,pp.708-728,sep.2013.
[6]p.m.molton,a.g.fassbender,andm.d.brown,“stors:thesludge-to-oilreactorsystem,”cincinnati,oh,1986.
[7]w.l.kranichanda.e.eralp,“conversionofsewagesludgetooilbyhydroliquefaction,”cincinnati,oh,1984.
[8]c.xuandj.lancaster,“conversionofsecondarypulp/papersludgepowdertoliquidoilproductsforenergyrecoverybydirectliquefactioninhot-compressedwater.,”waterres.,vol.42,no.6-7,pp.1571-1582,mar.2008.
[9]y.kalogoandh.monteith,“stateofscieneereport:encrgyand
resourcerecoveryfromsludge,”2008.
[10]l.nazari,z.yuan,s.souzanchi,m.b.ray,andc.(charles)xu,“hydrothermalliquefactionofwoodybiomassinhot-compressedwater:catalystsereeningandcomprehensivecharacterizationofbio-crudeoils,”fuel,vol.162,pp.74-83,2015.
[11]n.bradley,“theresponsesurfacemethodology(thesis),”indianauniversitysouthbend,2007.
[12]j.n.sahu,j.acharya,andb.c.meikap,“responsesurfacemodelingandoptimizationofchromium(vi)removalfromaqueoussolutionusingtamarindwoodactivatedcarboninbatchprocess,”j.hazard.mater.,vol.172,no.2-3,pp.818-825,2009.
[13]d.r.vardon,b.k.sharma,j.scott,g.yu,z.wang,l.schideman,y.zhang,andt.j.strathmann,“chemicalpropertiesofbiocrudeoilfromthehydrothermalliquefactionofspirulinaalgae,swinemanure,anddigestedanaerobicsludge.,”bioresour.technol.,vol.102,no.17,pp.8295-8303,sep.2011.
[14]americanpublichealthassociation(apha),“standardmethodsfortheexaminationofwaterandwastewater,”20thed.,washington,dc,usa,1960.
[15]c.tian,b.li,z.liu,y.zhang,andh.lu,“hydrothermalliquefactionforalgalbiorefinery:acriticalreview,”renew.sustain.ehergyrev.,vol.38,pp.933-950,2014.
[16]h.huang,x.yuan,h.zhu,h.li,y.liu,x.wang,andg.zeng,“comparativestudiesofthermochemicalliquefactioncharacteristicsofmicroalgae,lignocellulosicbiomassandsewagesludge,”energy,vol.56,pp.52-60,jul.2013.
[17]b.n.kuznetsov,s.a.kuznetsova,v.a.levdansky,a.v.levdansky,n.y.vasil’eva,n.v.chesnokov,n.m.ivanchenko,l.djakovitch,andc.pinel,“optimizedmethodsforobtainingcelluloseandcellulosesulfatesfrombirchwood,”woodsci.technol.,vol.49,no.4,pp.825-843,2015.
[18]g.shulga,s.vitolina,v.shekels,l.belkova,g.cazacu,c.vasile,andl.nita,“ligninseparatedfromthehydrolyzateofthehydrothermaltreatmentofbirchwoodanditssurfaceproperties,”cellul.chem.technol.,vol.46,no.5-6,pp.307-318,2012.
[19]z.daud,m.zainuri,m.hatta,a.sari,m.kassim,h.awang,a.m.aripin,v.education,u.tun,andh.onn,“analysisthechemicalcompositionandfibermorphologystructureofcornstalk,”vol.7,no.9,pp.401-405,2013.
[20]j.flandez,i.gonzález,j.b.resplandis,n.e.elmansouri,f.vilaseca,andp.mutjé,“managementofcornstalkwasteasreinforcementforpolypropyleneinjectionmouldedcomposites,”bioresources,vol.7,no.2,pp.1836-1849,2012.
[21]h.chen,q.han,r.a.venditti,andh.jameel,“enzymatichydrolysisofpretreatednewspaperhavinghighlignincontentforbioethanolproduction,”vol.10,no.3,pp.4077-4098,2015.
[22]l.zhang,p.champagne,andc.(charles)xu,“bio-crudeproductionfromsecondarypulp/paper-millsludgeandwastenewspaperviaco-liquefactioninhot-compressedwater,”energy,vol.36,no.4,pp.2142-2150,apr.2011.
[23]s.s.toor,l.rosendahl,anda.rudolf,“hydrothermalliquefactionofbiomass:areviewofsubcriticalwatertechnologies,”energy,vol.36,no.5,pp.2328-2342,may2011.
[24]s.yin,r.dolan,m.harris,andz.tan,“subcriticalhydrothermalliquefactionofcattlemanuretobio-oil:effectsofconversionparametersonbio-oilyieldandcharacterizationofbio-oil.,”bioresour.technol.,vol.101,no.10,pp.3657-3664,may2010.
[25]c.jazrawi,p.biller,a.b.ross,a.montoya,t.maschmeyer,andb.s.haynes,“pilotplanttestingofcontinuoushydrothermalliquefactionofmicroalgae,”algalres.,vol.2,no.3,pp.268-277,jul.2013.
[26]s.cheng,“bio-basedphenolicresinsandadhesivesderivedfromforestryrrsidues/wastesandlignin(thesis),”lakeheaduniversity,2011.
[27]d.r.vardon,b.k.sharma,g.vblazina,k.rajagopalan,andt.j.strathmann,“thermochemicalconversionofrawanddefattedalgalbiomassviahydrothermalliquefactionandslowpyrolysis.,”bioresour.technol.,vol.109,pp.178-87,apr.2012.
[28]t.matsui,a.nishihara,c.ueda,andm.ohtsuki,“liquefactionofmicro-algaewithironcatalyst,”vol.76,no.11,pp.1043-1048,1997.
[29]j.speight,handbookofpetroleumproductanalysis.2002.
[30]y.yang,a.gilbert,andc.(charles)xu,“productionofbio-crudefromforestrywastebyhydro-liquefactioninsub-/super-criticalmethanol,”aichej.,vol.55,no.3,pp.807-819,2009.
[31]p.sun,m.heng,s.sun,andj.chen,“directliquefactionofpaulowniainhotcompressedwater:influenceofcatalysts,”energy,vol.35,no.12,pp.5421-5429,2010.
[32]l.zhang,c.c.xu,andp.champagne,“energyrecoveryfromsecondarypulp/paper-millsludgeandsewagesludgewithsupercriticalwatertreatment.,”bioresour.technol.,vol.101,no.8,pp.2713-21,apr.2010.
[33]s.li,a.sanna,andj.m.andresen,“influenceoftemperatureonpyrolysisofrecycledorganicmatterfrommunicipalsolidwasteusinganactivatedolivinefluidizedbed,”fuelprocess.technol.,vol.92,no.9,pp.1776-1782,sep.2011.
[34]a.b.ross,p.biller,m.l.kubacki,h.li,a.lea-langton,andj.m.jones,“hydrothermalprocessingofmicroalgaeusingalkaliandorganicacids,”fuel,vol.89,no.9,pp.2234-2243,sep.2010.
- 还没有人留言评论。精彩留言会获得点赞!