零电荷电位基电容去离子的制作方法
发明背景
发明领域
发明领域是用于从溶液中除去盐和其它离子的电容(又称静电)去离子装置和方法。
定义
“吸附”是指将输入料流中的离子吸引到电极表面上并将这些离子留在电极表面上。
“amcdi”是指一种cdi池,其中各电极被膜包围并且其中阳极和阴极任一或两者含有表面电荷增强表面。
“amx-cdi”是指一种cdi池,其中各阳极被阴离子交换膜覆盖,而阴极保持未覆盖。
“amx-acdi”是指一种cdi池,其中各阳极被阴离子交换膜覆盖,而阴极未覆盖并含有负表面电荷。
“bet表面积”是指通过brunauer-emmett-teller法测定的表面积,其是使用氮气测定材料的表面积的物理吸附型方法。
“电容去离子”是指通过吸附从池的输入料流中除去离子并将去离子料流送往池输出。
“电容去离子池”是指利用静电力从输入料流中吸附离子的池。在“传统”或“常规”电容去离子池中,对阳极施加正电压并对阴极施加负电压,以在施加电压的同时使负离子吸附到阳极上和使正离子吸附到阴极上。
“池(cell)”是指暴露在输入料流下的多个电极,具有输出料流/废物料流的出口、连接到电极上的短路开关或电源,以及控制电源的装置。池可任选包括控制输入料流和输出料流/废物料流的装置。
“充电电位”是指施加到池电极上或是池电极的表面官能团中固有的并使池输入料流中的离子移向电极的电压。
“cmx-cdi”是指一种cdi池,其中各阴极被阳离子交换膜覆盖,而阳极保持未覆盖。
“cmx-acdi”是指一种cdi池,其中各阴极被阳离子交换膜覆盖,而阳极未覆盖并含有正表面电荷。
“电导率”是指输入料流、输出料流或废物料流的电导率。电导率是输出料流或废物料流中的离子的摩尔浓度的替代量度。电导率与此类料流中的离子摩尔浓度成正比。
“共离子”在cdi池中是指在阴极电位高于其epzc时被吸引到阴极上的阴离子和在阳极电位低于其epzc时被吸引到阳极上的阳离子。
“抗衡离子”是指被吸引到带正电荷的电极上的负离子和被吸引到带负电荷的阴极上的正离子。
“cx”是指碳干凝胶。cx电极具有标称表面积~200平方米/克的介孔结构。
“周期”是指电容去离子池的运行、吸附、接着解吸的一个周期。
“去离子”是指通过吸附到电极表面上而除去输入料流中的离子并作为输出物排出去离子料流。
“去离子池”是指从输入料流中除去离子的池。去离子池具有各种类型,例如传统的、mcdi、amcdi、i-cdi。
“解吸”是指从电极中释放吸附的离子到废物料流中。
“放电电位”是指施加到池电极上或是池电极的表面官能团中固有的使离子从电极解吸到废物料流中的降低或反极性电压。
“电极”是指导电的材料,通常多孔碳。
“i-cdi池”是指根据本文中公开的发明的“反向”电容去离子池。
“epzc”或“零电荷电位”是指在该电极电位下,表面处的离子吸附最小化。
“eo”是在电极短路时电容去离子池相对于参比电极的电位(即eo是短路条件过程中的电位)。
“流速”是指输入、输出或废物料流的通常以l/hr、ml/min等计的流速。
“输入料流”是指进入池的液体,通常是含有各种离子的水。
“mcdi池”是指一种cdi池,其中各电极被膜包围。
“膜”是指附着或施加到电极上的碳或碳基织物或涂层。
“n-”是指负表面电荷,例如n-cx是指具有带净负电荷的表面基团的碳干凝胶电极。
“输出料流”是指已经过吸附式去离子池并含有比输入料流中低的离子摩尔浓度的液体。
“p-”是指正表面电荷,例如p-cx是指带净正电荷的表面基团的碳干凝胶电极。
“phpzc”是指通过改变溶液的ph测定的在给定epzc下的溶液ph。
“极化窗口”是指用于进行电容去离子池的去离子(吸附)和再生(解吸)的电位/电压的跨度或范围。
“极性”是指dc电压的极性,正或负。
关于电极的“原始”是指没有表面改性;例如,如制造商供应的spectracarb电极是原始的。
“净化”是指从输入料流中除去离子。净化包括水软化,即除去硬水中的钙、镁和某些其它金属阳离子。
epzc的“重新定位(relocation)”是如循环伏安图中所示,由吸附/解吸周期的累积造成的电极的epzc的电位(又称“位置”)变化。
epzc的“位移”或“定位(position)”是指通过电极表面的有意的化学或电化学改性改变电极的epzc的电位(又称“位置”)。
“sc”是指spectracarb碳电极——例如在循环伏安法中常用作参比电极的碳电极。
“sce”是指饱和甘汞电极——一种标准参比电极。
“si-cx”是指二氧化硅涂布的碳干凝胶。
“表面电荷增强表面”是指已处理的电极表面。
“处理”是指改性电极表面以改变如本文中公开的电极的epzc。
“未处理”是指未经本文中公开的电极表面改性的电极,即原始碳电极。
“电压”和“电位”在本文中同义。除非另行规定,电压是直流(“dc”)的。
“废物料流”是指已经过解吸式去离子池并含有比输入料流中高的摩尔浓度的离子的液体。
“ζ电位”是分散介质和分散粒子周围的流体之间的电位差。
相关技术
随着全球人口增长和需水量持续增加,获得饮用水的途径也越来越重要。为达到用于食用、农业、发电厂或许多其它用途之一的水标准,必须满足许多水净化条件,包括盐含量水平。由于大部分盐的小分子尺寸,通过传统过滤方法难以分离盐含量。许多其它溶解的离子化合物经由典型的化学制备途径分离也具有挑战性。溶解离子如na+、k+、ca2+、fe2+、fe3+、cu2+、mg2+、cl-、so42-和no3-通常存在于水源中并且需要特定分离方法以产生净化水。传统分离方法包括多级闪蒸(msf)和反渗透(ro)。在msf中,使用蒸馏法从盐水料流中分离水,其中将水煮沸并收集蒸气以产生纯净水流。尽管有效,但这种方法相当能量密集并受困于设备腐蚀问题。在ro中,使用极小孔径以将略小的水分子与较大水合盐分子分离。这种方法需要压力梯度以克服盐溶液的渗透压和使水跨过半透膜传送到纯化渗透料流中。尽管ro通常比msf更有效,但其需要能够实现更高压力的泵并发生膜表面的有机、生物和沉淀基结垢,这最终限制该分离法的寿命并提高支出,尤其是在市政规模下。
电容去离子(cdi)是一种新兴分离法,其依赖于利用静电学从水/水溶液中分离溶解盐。在传统cdi中,向一对(通常)碳电极施加电位,阳极被定义为被施加正电位的电极,阴极被定义为被施加负电位的电极。在这种传统cdi法中,如图1中所示,负离子或阴离子,如cl-和so42-被吸引到正向极化的电极(阳极)上,而正离子或阳离子,如na+和ca2+被吸引到负向极化的电极(阴极)上。当碳电极被盐/离子饱和时,降低、短接、移除或逆转(手动或在计算机控制下)外加电位以将这些离子解吸到浓缩废物料流中。当降低、短接或移除池电位时,在碳表面处用于离子吸附的驱动力随后降低,以造成离子解吸。解吸(从电极表面除去离子)使碳再生以在向电极再施加电压后用于吸附/分离更多离子(如下文解释,电极表面的解吸在传统cdi中不完全)。为加速解吸,可以反转向电极施加的电压极性:电极表面上的反极性(负极性-阳极,正极性-阴极)会排斥共离子,以造成与非反向极性的开路、短路或降低的电压相比更快的离子解吸。用于吸附的初始外加电位通常为0-2.0v,但已经报道高达3.0v的值。由此可以从溶液中定期除去盐并解吸到浓缩废物料流中。
用传统cdi分离水性氯化钠
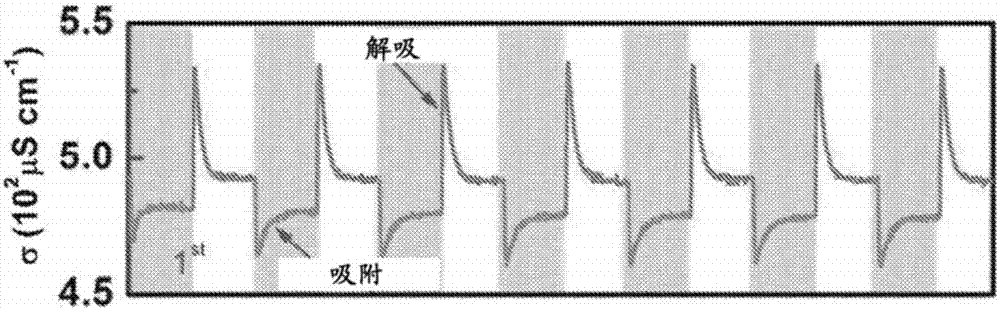
尽管这种方法听起来相对简单,但实际上,该分离法更复杂,例如改变表面电极性质显著改变该盐分离法。cdi法中所用的碳电极通常被设计为惰性、高表面积(即非常多孔)和导电的电极以在通过外加电位调节的同时吸附和解吸离子。向碳电极施加电位改变大部分碳电极的性质。如图2中所示,最初,盐在外加电位下吸附在cdi池中(阴影区),输出料流的电导率随外加电位而降低(输出料流的电导率的降低意味着总离子吸附增加),然后在池短路时解吸(非阴影区)。在施加电压时存在峰值(在施加电位时初始尖峰至最低σ(电导率),显示在图2的阴影区中)并在移除电压时存在反向峰值(或逆转或降低,如图2中的电极短路时尖峰至最高σ(电导率)所示,显示在图2的非阴影区中);图2显示交替向电极施加充电电位(阴影区),然后电极短路(非阴影区))。使用~4克碳干凝胶电极、1.5毫米有机硅隔片、2升4.3mmn2脱气nacl溶液、各30分钟的吸附和解吸时间、1.2v的充电电位、0v的放电电位(短路)和75mlmin-1的流速进行这一实验。图2显示cdi法的前7个周期。尽管最初稳定,随着反复循环,这一吸附-解吸行为开始变化:施加充电电位后的较高σ(电导率的降低较低)意味着吸附少得多的离子。图3中显示在227个周期后cdi池的脱盐响应。在227个周期后,显而易见,在存在和不存在外加电位时电导率的稳态浓度差体现在基本平坦的电导率σ曲线中:电导率的变化已降至几乎0,即在阴影(施加1.2v)和非阴影(0v,短路)区中,稳态电导率或盐含量水平都几乎相同。图3中所示的在外加电压下的逆转峰(inversionpeak)表明该分离法的失效,这导致从输入料流中净脱盐降低。这意味着该cdi池不再充当盐分离装置:其已达到装置寿命的终结并且必须更换。当对照周期数绘制盐吸附容量(以盐的毫克数/克碳电极计)时,结果显示在图4中。盐吸附容量γ在方程1中定义为:
γ=(δσmv)/(mc)(1)
其中σ是存在和不存在外加电位时的电导率差异,m是分子量,v是溶液体积,m是碳电极的质量,且c是盐浓度vs.电导率的校准常数。在图4中清楚可见,碳电极吸附盐的盐吸附容量不断衰减直至看不出分离。这一衰减过程不是这一实例中所用的碳独有的,也已对碳干凝胶、spectracarb活性炭布和
λ=(γf/m)/(qad)(2)
其中γ是盐吸附容量,f是法拉第常数,m是分子量,且qad是在充电过程中传递的总电荷。在图5中显示随着盐吸附容量降至0电荷效率的损失。为了将静电分离(又称电容去离子)法商业化,要求显著提高池寿命。在汽车之类中,cdi池使汽油里程数变糟并且在经济上不可接受。如果cdi池以10天的现行cdi池寿命运行,并且如果各cdi池花费$5000,反映每10天更换池的2年池更换成本是$365,000(但不包括人工和管理成本)——明显不可接受的值。cdi池寿命取决于总周期,因此实际cdi池寿命比10天短得多。
碳电极中的研究集中于改进用于构造超级电容器电极的碳材料的孔隙率,但尚未进行特别与离子分离和水净化相关的研究。传统cdi系统的已知进展包括新的池设计、不对称电极涂层和离子交换膜的应用。在本文中公开的本发明之前已知的电容去离子技术中的最成功进展可能是添加离子交换膜以形成膜电容去离子(mcdi)。mcdi池不仅提供更可靠的分离方法,还提供更高的盐吸附容量。但是,将膜添加到传统cdi池中显著提高总池成本,以致在大规模下的商业成功困难得多,并且没有解决图4中所示的传统cdi池潜在的累积(累积周期依赖(aggregatecycledependent))衰减过程(即短池寿命,又称失活)
发明概述
要解决的技术问题是提供解决传统电容去离子(cdi)和膜电容去离子(mcdi)装置和方法的短寿命问题的电容(又称静电)去离子装置和方法。本文中公开的解决方案(i)正确表征之前误解的电极epzcs的累积重新定位和(ii)提供极长寿命电容去离子装置和方法的电极表面改性。不同于现有技术装置,本文中公开的“反向电容去离子”(“i-cdi”)装置的电极表面在每次解吸至最低离子条件后复原(即吸附性基于与第一使用周期一样),由此提供具有极大改进的分离寿命的电容去离子池。此外,所公开的方法的放电阶段可通过i-cdi池中的电容放电生成电力。获自长期cdi试验的结果证明在静电基分离池的构造中获得定向和稳定的表面化学的重要性。
本发明人在本文中首次公开了在稳定的盐分离法的构造中使用预处理过的氧化碳阳极的静电分离法(又称电容去离子法),由此减轻在之前的cdi和mcdi装置和方法中看出的衰减和短池寿命问题及其它实施方案。本文中公开的本发明在评估和改进电容去离子池和电容基离子分离法的容量、电荷效率和池寿命中利用“电极表面电荷”技术的改进,尤其是定位(又称“移动”)“零电荷电位”(epzc)。本文中公开的本发明提供在比对现有cdi和mcdi池所见明显更长的时间内具有盐分离效率的池。本发明人的研究集中于(i)电极表面改性化学以改进用于离子分离和水净化的电极的电荷存储容量和电荷效率,和(ii)如何和在多大程度上移动阳极和阴极的epzc。除本发明人的公开外,还没有关于作为一种改进电容去离子技术的手段改变电极表面电荷的研究。
本发明的第一实施方案包含一种结构,其包含至少一个入口、至少一个出口、至少一个阳极、至少一个阴极、操作以向至少一个阳极和至少一个阴极施加短路或用户可选的dc恒定电压或恒定电流的开关,和经所述入口进入和经所述出口排出的离子溶液,所述离子溶液通过与至少一个阳极和至少一个阴极接触而去离子,其中已通过阳极表面的改性使至少一个阳极的epzc的位置移向提高的epzc。所述阳极改性来自选自通过暴露在酸下氧化、共价连接在与所述离子溶液接触并且未向阳极施加电压时带负电荷的官能团、将二氧化硅官能团共价连接到阳极上、连接磺酸基团和连接可造成离子溶液中的负ζ电位的任何表面官能团的处理。
本发明的第二实施方案包含一种结构,其包含至少一个入口、至少一个出口、至少一个阳极、至少一个阴极、操作以向至少一个阳极和至少一个阴极施加短路或用户可选的dc恒定电压或恒定电流的开关和经所述入口进入和经所述出口排出的离子溶液,所述离子溶液通过与至少一个阳极和至少一个阴极接触而去离子,其中已通过阴极表面的改性使至少一个阴极的epzc的位置移向降低的epzc。阴极的改性来自选自通过暴露在n2、ar和h2的还原处理下还原、共价连接在与所述离子溶液接触并且未向阴极施加电压时带正电荷的官能团、将胺官能团共价连接到阴极上、还原的碳表面(包括碳基面(basalplane))、连接氧化铝表面物类(species)和连接可造成离子溶液中的正ζ电位的任何表面官能团的处理。
本发明的第三实施方案包含一种结构,其包含至少一个入口、至少一个出口、至少一个阳极、至少一个阴极、操作以向至少一个阳极和至少一个阴极施加短路或用户可选的dc恒定电压或恒定电流的开关和经所述入口进入和经所述出口排出的离子溶液,所述离子溶液通过与所述至少一个阳极和至少一个阴极接触而去离子,其中已通过阳极表面的改性使至少一个阳极的epzc的位置移向提高的epzc,且其中已通过阴极表面的改性使至少一个阴极的epzc的位置移向降低的epzc。阳极的改性来自选自通过暴露在酸下氧化、共价连接在与所述离子溶液接触并且未向阳极施加电压时带负电荷的官能团、将二氧化硅官能团共价连接到阳极上、将磺酸基团连接到阳极上和由在o2环境中加热或经电化学方式的氧化处理的处理。其中阴极的改性来自选自通过暴露在n2、ar和h2的还原处理下还原、共价连接在与所述离子溶液接触并且未向阴极施加电压时带正电荷的官能团、将胺官能团共价连接到阴极上、减少包括碳基面的碳表面、和在离子溶液中具有正ζ电位的材料的处理。
在所有实施方案中,阳极、阴极和离子溶液包含在一种结构中,所述结构具有经其引入溶液(输入料流)并允许其接触阳极和阴极的入口、穿过该结构的电导体穿透(penetrations)以允许连接到阳极和阴极上的短路开关工作或向阳极和阴极施加用户可选电压,和经其从该结构中排出去离子溶液(输出料流)或含解吸离子的溶液(废物料流)的该结构中的出口。该结构通常除入口、出口和电导体穿透外是封闭的以实现阳极和阴极的短路或向阳极和阴极施加电压。该结构及其内容物被称作去离子池。在另一些实施方案中,可以无线控制该开关和内部电源以避免该结构被电导体穿透。
输入料流的净化(去离子)程度主要由给定体积的输入料流接触的池中的总电极表面积、解吸前用于离子吸附的暴露持续时间、在该系统中调制的电压(电位)或电流和工作电压窗口(下述)控制。通过由该去离子池供应或提取的电流的调制或通过由该去离子池供应或提取的电压的调制控制净化(去离子)程度能够对去离子细调。
本发明的实施方案可用于净化发电厂废水、发电厂冷却水、洗衣废水、为供人类食用净化的水、为农业净化的水、为园艺净化的水、为用于食品净化的水、要软化的水、为供人类食用净化的海水、为实验室用途净化的水、为人类食用或农业应用净化的微咸水和为医疗用途净化的水。
附图简述
图1.电容去离子(cdi)过程,其中盐在外加电位(在此由电源供应)的影响下吸附/脱除并在降低、短接、移除或逆转电位时解吸。图1的右侧上的池表现出阳极和阴极的短路,以使离子从电极解吸。
图2.在1.2/0v下循环7个周期的cdi池的吸附(阴影)和解吸(非阴影)。波动的信号表明在向电极施加电位然后短路时的反复吸附(阴影区,1.2v)和解吸(非阴影区,0v)事件。
图3.图2中所示的cdi池在1.2/0v下227个周期后的反复循环。现在在吸附和解吸步骤之间看出最小电导率差异。
图4.在1.2/0v下循环的cdi池的盐吸附容量γ
图5.在1.2/0v下循环的cdi池的电荷效率λ。
图6.原始碳干凝胶电极在n2脱气4.3mmnacl溶液中在1mv/s下的循环伏安图(cv)。
图7.原始碳干凝胶电极以及用作cdi池中的正极(阳极)和负极(阴极)的碳干凝胶电极的循环伏安图(cv)。这些试验在n2脱气4.3mmnacl溶液中在1mv/s下进行。
图8.根据图7的用过的电极的电位分布。图中所示的阳极处的epzc(epzc+)和阴极处的epzc(epzc-)已基于伏安图中的阳极和阴极epzcs平均化。e+和e-分别是向阳极和阴极施加的电位。
图9a和9b显示用硝酸氧化的碳干凝胶(cx)样品的ftir(图9a)和表面酸度分析(图9b)。
图10a和10b显示碳干凝胶(图10a)和sc电极(图10b)的cv,表明通过硝酸处理使epzc定位在更正性的电位。
图11a和11b显示与反向电容去离子(i-cdi)池(图11b)相比传统cdi池(图11a)的运行,其中在短路电位下发生离子吸附并在外加电位下发生解吸。
图12.在左侧,当这两个电极的epzcs相对于短路电压(eo)不同地定位时,阳极和阴极处的epzcs之间的电位窗口可用于脱盐,但吸附-解吸行为反转。在右侧,当在四电极池中使用si-cx阳极和原始cx阴极时,在4.3mm脱气nacl溶液中在0.8和1v的总池电位下电位在阳极(e+)和阴极(e-)处分布。
图13a和13b显示在31升4.3mm脱气nacl溶液中在75mlmin-1下i-cdi和cdi系统在0.8/0v下的初始周期(第3-第5)的电导率(σ)(图13a)和电流密度(j)(图13b)。
图14a和14b显示在31升4.3mm脱气nacl溶液中在75mlmin-1下i-cdi(图14a)和cdi系统(图14b)在0.8/0v下的第50-57周期的电导率的选定的廓线(profile)。
图15a、15b和15c显示在0v下放电的i-cdi和cdi系统的放电步骤的盐吸附容量(γ)(图15a)、传递的电荷(q)(图15b)和电荷效率(λ)(图15c)。此外,将在1.2/0v下使用的cdi系统的数据添加到曲线图中以供比较。
图16a、16b和16c显示在0.8v下充电的i-cdi和cdi系统的放电步骤的盐吸附容量(γ)(图16a)、传递的电荷(q)(图16b)和电荷效率(λ)(图16c)。此外,将在1.2/0v下使用的cdi系统的数据添加到曲线图中以供比较。
图17.通过与cx阴极和si-cx阳极一起使用的i-cdi系统增强的稳定性。在31升4.3mm脱气nacl溶液中在0.8/0v下进行这一试验。显示与使用原始cx电极在类似条件下的标准cdi运行的性能比较。在这一曲线图中,已添加回归得到的线。
图18a、18b和18c显示在空气/氧气中的热处理(图18a)、酸处理以氧化碳表面(图18b)和使用teos的二氧化硅涂布法(图18c),它们都产生具有正向移动的epzcs(负表面电荷)的碳电极。
图19a和19b显示碳干凝胶(cx)电极的原硅酸四乙酯(teos)处理以在碳干凝胶电极上产生二氧化硅基团(cx/teos)以使epzc正向移动(图19a),和市售spectracarb(sc)的硝酸处理以在碳干凝胶电极上产生硝酸根基团(sc/hno3)以使epzc正向移动(图19b)。
图20.使用spectracarb电极的乙二胺处理的碳表面的胺官能化。
图21a、21b和21c显示原始和处理过的sc的化学表征。样品的傅里叶变换红外(ftir)光谱显示在图21a中。这些样品使用具有不同ph值的4.3mmnacl溶液进一步测试以评估零电位点(phpzc)的值,并显示在图21b中。在4.3mm脱气nacl溶液中在0.5mvs-1下进行电极的循环伏安图并显示在图21c中。通过电流密度除以电压扫描速率计算电容。phpzc和零电荷电位(epzc)值在图21b和21c中用箭头强调。
图22a和22b通过浓度(图22a)和通过电流密度(图22b)显示在用16片p-sc阴极和16片n-sc阳极配置i-cdi池时的选定周期。在~31升~4.3mm脱气nacl溶液中在20mlmin-1下在用于盐解吸的不同充电电压和用于盐吸附的短路电压下进行这些试验(x/0v,其中x=0.15-1.25)。各充电和放电半周期花费4000秒。
图23a和23b显示对于配有表面电荷增强sc和cx电极的i-cdi池用于盐解吸的充电步骤(图23a)和用于盐吸附的短路(图23b)的性能评估。cx*和sc*指示配有原始阴极代替p-cx和p-sc阴极的i-cdi池。
图24a和24b显示膜电容去离子(mcdi)过程,其中盐在外加电位(在此由电源供应)的影响下吸附/脱除(图24a)并在降低、短接、移除或逆转电位时解吸,其中将阴离子交换膜置于阳极上并将阳离子交换膜置于阴极上(图24b)。
图25a和25b显示具有正表面电荷增强阳极和负表面电荷增强阴极的不对称膜电容去离子(amcdi)。图25a显示在向阳极和阴极施加电位时的吸附。图25b显示在阳极和阴极在0v电位下时的解吸。
图26a和26b显示cdi和mcdi性能的比较。电导率廓线(图26a)和放大电导率廓线(图26b)。在用碳干凝胶电极形成的分批模式布置中在600毫升5mmnacl溶液再循环下在1.2/0v下循环运行。
图27a和27b显示cdi和mcdi性能的比较。经测试期的充电电流(图27a)和电吸附容量(图27b)。在用碳干凝胶电极形成的分批模式布置中在600毫升5mmnacl溶液再循环下在1.2/0v下循环运行。
图28a和28b显示在阳极(图28a)和阴极(图28b)处用过的cdi碳干凝胶(cx)电极的零电荷电位(epzc)的后测量。
图29a和29b显示在阳极(图29a)和阴极(图29b)处用过的mcdi碳干凝胶(cx)电极的零电荷电位(epzc)的后测量。mcdi抑制epzc的位移。
图30a和30b显示配有原始和氧化cx电极的组合的cdi(图30a)和mcdi(图30b)池的性能。在用碳干凝胶电极形成的分批模式布置中在600毫升5mmnacl溶液再循环下在1.2/0v下循环运行。
图31a和31b显示配有原始和氧化cx电极的组合的mcdi池的电子电荷性能(图31a)和放大电流廓线(图31b)。在用碳干凝胶电极形成的分批模式布置中在600毫升5mmnacl溶液再循环下在1.2/0v下循环运行。
图32a、32b和32c显示配有用作阳极(图例“a”)和阴极(图例“c”)的原始(pr)和氧化(ox)电极的组合的mcdi池的性能结果概要,包括传递的电荷(图32a)、盐吸附容量(图32b)和电荷效率(图32c)。
图33a和33b显示原始(pr)和氧化(ox)
图34a和34b显示用原始(pr)和氧化(ox)
图35a和35b显示用原始(pr)和氧化(ox)
图36a和36b显示spectracarb(sc)电极的氮气吸附(图36a)和原始和氧化sc电极的epzc位置(图36b)。
图37a、37b、37c和37d显示具有spectracarb电极(sc)的mcdi和amcdi池的性能,包括电导率(图37a)、盐吸附容量(图37b)、电荷效率(37c)和传递的电荷(图37d)。在分批模式布置中在1000毫升5mmnacl溶液再循环下在1.2/0v下循环运行。
图38a、38b和38c显示用原始spectracarb(sc)阳极和阴极电极形成的cdi、mcdi、仅阳离子膜cdi(cmx-cdi)和仅阴离子膜-cdi(amx-cdi)池的电导率(图38a)、溶解氧(图38b)和ph(图38c)廓线。在分批模式布置中在1000毫升5mmnacl溶液再循环下在1.2/0v下循环运行。
图39a、39b、39c和39d显示用具有原始sc阳极和阴极电极的mcdi池和用原始sc阳极和氧化sc阴极电极形成的仅阴离子膜-不对称cdi(amx-acdi)池的电导率(图39a)、溶解氧(图39b)、ph(图39c)和电流(图39d)廓线。在分批模式布置中在1000毫升5mmnacl溶液再循环下在1.2/0v下循环运行。
图40a、40b和40c显示传统mcdi、amcdi、cdi和单膜cdi池的电吸附容量(图40a)、电子电荷(图40b)和电荷效率(图40c)。
图41.基于分别$5,000、$10,000、$5,000和$7,500的资本和更换成本,cdi、mcdi、i-cdi和amx或cmxacdi的预计资本和更换成本。分别使用10、180、365和180天的cdi、mcdi、i-cdi和amx或cmxacdi的装置寿命。
图42.在cdi(优先阴离子吸附)和i-cdi(阳极和阴极处的离子吸附没有显著差异)池中存在和不存在外加电位下的ph波动。cdi池表现出较大ph波动,其中使用两个类似的表面电荷增强电极。
图43.通过制造氢氧化物(较高ph)和带电电极而将硼转化成硼酸盐以从溶液中除硼。该溶液的ph可以通过表面带电电极或通过溶解物类的还原/氧化调节。
优选实施方案详述
在图6中显示原始碳干凝胶电极在n2脱气4.3mmnacl溶液中在1mv/s扫描速率下的循环伏安图(cv)。cv与线性扫描伏安法的区别在于在cv实验中达到设定电位后,工作电极的电位朝相反方向升高以回到初始电位。对照外加电压(即工作电极的电位)绘制工作电极处的电流以提供循环伏安图迹线。该cv迹线通常滞后,甚至在完美可逆机制下。甚至可逆couples也含有极化过电位并因此在电位从负升至正然后从正降至负(经过初始电位)时表现出滞后迹线。这种过电位源自被分析物(例如离子)扩散速率和从电极向被分析物传递电子的固有活化势垒的组合。不同于对外观上大多为盒状的超级电容器看出的大多数电容曲线,在更稀电解质中的cv具有明显更多“特征(feature)”。本发明人首次认识到可以利用这些特征,特别是零电荷电位和解吸的关系改进电容去离子。在图6中显示通过“pzc”标注的峰和谷(在本文中是指“epzc”)。峰和谷是指零电荷电位epzc的位置,其中电极具有电荷存储或电容的最小值。如果向电极施加电位以到达这一epzc区,该电极具有离子吸附的最小值。在图6中,在短路电位的右侧,促进正极上的阴离子吸附,而在负极上的pzc的左侧,促进阳离子吸附。因此,在以去除离子为目标的电容去离子池中,epzc的位置对离子的有效吸附至关重要;之前没有认识到,epzc的位置对离子的解吸也至关重要。
在图6中也显示cdi池的短路电位的位置。这一位置是在将池放电至0v(阳极短接到阴极上)时电容去离子池恢复到的电位。在电源是传统dc电源或蓄池的cdi池中,将正端子连接到阳极上,并将负端子连接到阴极上。在向cdi池施加电位时,阳极(正极)处的电位会变得更正,而阴极(负极)处的电位会变得更负。对于阳极,当向阳极施加正电位时,电位会从在短路电位处的位置提高到在由右侧的虚线灰框区突显的区域中的某处。由于这些电位都比epzc更正,仅促进阴离子吸附。在阴极处,表现出相反的情况。当向这一电极施加负电位时,电位会从在短路电位处的位置降低到在由图中的虚线灰框或实线灰框突显的区域中的某处。由于epzc位于比这一池中的短路电位更负的电位,在发生抗衡离子吸附之前必须从碳表面排出共离子。在阴极处的这种共离子排出过程(即在阳极和阴极短接时阴离子被正性阴极电位吸引)导致在使用原始和未处理的碳电极时该分离法中的低效率。但是,在超过1.0v的大多数标准电位下,在传统cdi法中发生净脱盐。之前未解决的技术问题之一是为何cdi分离如图4中的结果所示在短时间内(运行25-100小时,假设30分钟周期)内失效。现在通过考察cdi池的循环伏安图回答这一问题。在图7中显示原始和用过的碳干凝胶电极的叠加cv。用过的碳干凝胶电极来自图4中描绘的cdi实验中的正电极(阳极)和负电极(阴极)。显而易见的是负电极(即阴极,向其施加负充电电位)的cv看起来非常类似于原始电极,表明在该电极的表面上几乎没有改变。还显而易见的是,负电极的epzc位于与原始电极类似的位置。但是,正电极(阳极)的情况下明显不同。epzc已明显朝正向重新定位,表明阳极表面的化学组成永久改变。使用与图6类似的论点,正电极处的epzc的这种重新定位现在导致cdi法的失效增加。当在短路后在用过的cdi池的正和负电极处施加充电电位时,这两个电极都排出共离子以及吸附抗衡离子。这种混合吸附/解吸过程导致从输入料流中净脱除离子降低,由此降低盐吸附容量和电荷效率。图5和图7中标注的“j”是碳电极的以ma/g计的电流密度。在这些图中,e+是向阳极施加的电位,e-是向阴极施加的电位,且eo是该cdi池在短路下的电位。在cdi运行过程中的epzc重新定位提供解释在1.2/0v下运行过程中发现的性能损失的关键见解。被斜杠分开的电压是指在该斜杠左边的充电电位和在该斜杠右边的放电电位。1.2/0v充电和放电电位产生图8和图7中所示的分布,并反映用过的cdi池中的epzcs。如所提到,在1.2/0v下的池运行过程中,由于这两个电极处的吸附/解吸的累积失效(cumulativeinefficiencies),观察到离子(尤其是na+和cl-)吸附容量的衰减。该失效最好解释为两个冲突的驱动力,即一个有助于阴离子解吸,另一个有助于阳离子解吸。由于η*-,即用于阴离子解吸的驱动力(即η*-=eo-epzc-,eo是cdi池的短路电位),已存在于与原始电极相关的电位分布中(图6中的共离子排出)并且甚至在1.2/0v下长时间运行后也保持这一驱动力(图7),在阴极处保持失效;但是,η*+,用于阳离子解吸的驱动力的形成(即η*+=epzc+-eo)和cdi池衰减的潜在成因,与阳极的epzc跨过eo电位重新定位相关联(即原始阳极的epzc略负,但用过的阳极的epzc非常正)。基于我们的电位分布和循环伏安研究,这一结论清楚显示在图8中。
在评估epzc的这种重新定位,尤其是阳极epzc的重新定位背后的原因时,可以使用各种技术。图6和图7中所用的cv是用于检测表面变化的相当灵敏的方法,但这种方法不能确凿地识别对此处进行的cv试验中epzc的这种位移负责的特定表面物类。因此,使用其它表面分析方法分析epzcs的重新定位,包括傅里叶变换红外光谱学(ftir)和表面酸度分析。在图9a和9b中显示用硝酸处理以将碳表面氧化的碳干凝胶电极的ftir和表面酸度分析。显而易见在硝酸处理后ftir谱中的表面氧化物基团和碳样品的表面酸度提高。在图10a和b中,显示碳干凝胶和spectracarb(sc)电极的cv,这证实在硝酸中氧化后epzc的正向重新定位。这些结果证实传统cdi池中的分离性能损失背后的化学/物理原因主要归因于碳阳极(正电极)的氧化。最终,为获得cdi池中的稳定分离,必须在阳极和阴极处都控制epzcs的位移,氧化是对阳极的主要担心。
图11a和11b比较传统和反向电容去离子法。在传统cdi中,如上所述,盐(或其它类型的离子)在外加电位的影响下吸附到通常碳电极上并在移除、短接、逆转或降低电位时解吸。在本文中公开的反向电容去离子(“i-cdi”)装置和方法中,逆转充电模式:盐在外加电位下(其中利用外加电位达到阳极和阴极处的epzcs)解吸并在移除、短接、逆转或降低电位时吸附。在阳极(正电极)和阴极(负电极)处都利用表面带电(即表面改性)电极使这一操作成为可能:由于化学改性的电极表面,阴离子通过使用正表面电荷增强的电极优先吸附在阴极处且阳离子通过使用负表面电荷增强的电极优先吸附在阳极处。在一个实施方案中,如图11b中所示,阳极可以由氧化或二氧化硅处理的碳电极构成,而阴极可以由原始或胺处理的碳电极构成。可以利用其它电极表面改性化学移动或定位阳极和阴极的epzcs,但本文中公开的表面改性化学是一些最经济和可预测的方法。
由于图11b中对i-cdi描绘的反向运行流程,定义i-cdi池的工作电压窗口,又称可用电压范围是重要的。通过阳极和阴极处的epzcs差异显示i-cdi法的工作电压窗口。可以利用epzc位置的这一差异在吸引离子所需的电位远离未处理的电极的阳极和阴极处的epzcs时从溶液中吸附阴离子和阳离子。内部工作电压窗口越大,可用于离子吸附/解吸的驱动力越大,这能够由于各电极处的较高盐吸附容量实现较小的商业装置尺寸。工作电压窗口的大小有效决定碳电极的最大盐吸附容量(γ)。
在图12中显示由二氧化硅处理的碳干凝胶阳极和原始碳干凝胶阴极构成的i-cdi池的工作电压窗口。对于这一实施方案,当阳极处的epzc为~0.62v且阴极处的epzc为~-0.17v时,工作电压窗口为大约0.8v(0.62-(-0.17)=0.79v)。在图12中的右侧是匹配它们各自的epzcs的位置并由此能够最大限度利用这一池中可提供的工作电压的分布电极电位(如通过4电极研究测得,其在向这一i-cdi池施加~0.8v时在阳极和阴极处分别显示e+和e-)。当池短路(eo)时,由于各电极处的电位远离它们各自的epzcs,离子吸附在i-cdi池中。尽管这一示例性池显示0.8v的工作电压窗口,但可以通过在阳极处制造(通过表面改性)具有更正性epzcs的电极材料和/或在阴极处制造具有更负性epzcs的电极材料而为反向吸附/解吸性能进一步扩大工作电压窗口。下面提出朝负方向的epzc位移或定位以增大这一方法的工作电压窗口。小至0.4v的工作电压窗口可有效将输入料流去离子;但是,如下文论述,工作电压窗口越大,本文中公开的本发明的实施方案的吸附容量越大。
为了演示i-cdi运行,用二氧化硅改性的阳极和原始碳阴极构造池。构造在阳极和阴极处都使用原始碳电极的相同池以供比较。在图13a和13b中显示在0.8/0v下运行的i-cdi和cdi池对该池的充电/放电的电流和电导率响应。在图13中显而易见当两者都暴露在相同摩尔浓度的含盐输入料流下时在与传统cdi比较时i-cdi池的反向电导率(吸附/解吸)性能。在i-cdi池中,盐在0.8v的外加电位下解吸(电导率提高)并在池短路或放电时吸附(电导率降低)。此外,在0.8v下在i-cdi中传递的电荷(“q”)比传统cdi情况中少,以带来总体更高的电荷效率(λ)。较低q意味着池更有效,即从输入料流中除去每摩尔离子的电子“用量”较低。在图14a、14b、图15a、15b、15c、16a、16b和图16c中显示为了充电/放电在0.8/0v下运行的i-cdi(图14a和如图15和16中标示)和cdi池(图14b和如图15和16中标示)和在1.2/0v下运行的cdi池的选定性能特征vs运行小时数,每个周期的吸附和解吸分钟数相同。
为了检查i-cdi过程的长期稳定性,i-cdi池运行600小时,电位在充电(0.8v)和放电(0v)之间循环。在图17中显示与为了充电/放电在1.2/0v和0.8/0v下运行的cdi池相比i-cdi过程的循环稳定性。i-cdi过程显示在此处测试的条件下≥500%的改进的寿命,证明当使用不同表面电荷增强的电极,例如显著减轻传统cdi中固有的现有性能衰减的有意氧化阳极(正电极)时这一过程的稳定性。
i-cdi工作电压窗口的扩大
为了提高i-cdi法的通用性,可以通过将工作电压窗口扩大到超过0.8v而获得更高的盐吸附容量(γ)。可以通过朝正方向提高阳极(正电极)处的epzc和/或朝负方向降低阴极(负电极)处的epzc实现这种扩大。有意地通过本文中公开的碳处理实施这些定向移动,处理程度与epzc的适当定位相关联。碳表面的氧化有助于正向移动epzc,碳表面的还原(减少氧化物基团数)有助于负向移动epzc。图10a中显示的数据表明,通过在较高温度下的硝酸处理提高碳表面的氧化会逐步正向移动epzc。还原处理同样造成负方向上的类似效应。除这些简单的氧化/还原处理外,表面涂层也对电极的表面电荷和所造成的其epzc的位置(这划定在i-cdi法中的工作电压窗口)起到关键作用。
为了验证表面基团对epzc位置的影响,在本发明人的实验中使用硝酸和二氧化硅表面涂层。在图18a至18c中显示三种处理方法(图18a(热处理),图18b(酸处理)和图18c(用二氧化硅膜涂布))以正向移动epzc的位置。电极化学领域中已知的其它方法可用于实现类似位移,包括在电极表面上产生带负电荷的官能团的任何处理,如此处显示的用于添加表面氧化物和二氧化硅基团的那些。在图19a和19b中显示原硅酸四乙酯(teos)和酸处理方法以正向移动两种碳电极的epzc:介孔碳干凝胶(cx)和微孔spectracarb(sc)。
在图20中显示使用乙二胺使碳电极被胺表面基团官能化的处理方法的一个实例。这种处理方法产生-nh3+表面基团、正向位移的phpzc和负向位移的epzc(如图21c中所示)。当这些胺官能化阴极碳电极(p-sc)与具有正向位移的epzcs(通过氧化处理或表面官能团,即n-cx或n-sc)的阳极碳电极组合时,可以如图12中所示将i-cdi法的工作电压窗口扩大到超过0.8v。当使用氧化/二氧化硅处理的碳阳极和原始碳阴极时,i-cdi法的原始工作电压窗口为~0.8v。
本发明人已经证实,在图7中所示的传统cdi实验以及图12中所示的i-cdi法中阳极epzc都朝正方向位移,在图21a-21c中标作n-cx和n-sc的电极是带负表面电荷的电极。使阴极epzc朝负方向移动以增大i-cdi法中的工作电压窗口的类似方法使用乙二胺进行胺处理以在碳表面上制造胺官能团,其在水溶液中带正电荷,以产生负向位移的epzc(在图21a-21c)中标作p-cx和p-sc)。如图22a-22b和23a-23b中所示,当原始阴极被胺官能化的碳(p-cx或p-sc)替代时,工作电压提高到≥1.0v。此处显示的数据是针对胺处理的碳阴极,但可以负向移动epzc(通过制造正表面电荷)的任何表面基团都会增强用于i-cdi法的电压窗口。
对于碳基静电分离,碳电极或任何电极的epzc的作用对成功分离(从输入料流中吸附离子)而言是基本的。在迄今显示的的实例中,cdi池的累积碳氧化已表明造成正向位移的epzc,这导致传统cdi的脱盐容量降低。当通过氧化或其它表面官能团(如二氧化硅基团)有意地使碳阳极的epzc朝正方向移动时,这一电极可以与具有负向位移的epzc(通过还原或其它表面官能团,如胺基产生)的阴极配对以制造反向电容去离子池,其中与传统cdi池相比,分离性能不随累积吸附/解吸周期衰减。接着,显示epzc对更复杂的电容去离子系统的影响,进一步证实这一参数在静电分离和通过本公开的发明实现的改进中的重要性。
与cdi相比,如果i-cdi池具有365天的分离寿命(与之前显示的传统cdi的10天相比)并且该单元的初始和更换成本为$5000,这相当于$10,000的2年成本,因为该单元需要每年更换,与具有$365,000的估算2年成本的传统cdi相比是明显的改进。
不对称膜电容去离子
如图24a-24b和25a-25b中所示的膜电容去离子(mcdi)是传统cdi的改进,其稍微减轻使用传统cdi池所见的降低的盐吸附容量和电荷效率。关于由外部电源向池电极施加电压,mcdi池的运行与cdi池的运行相同。cdi和mcdi池之间的结构差异是在池中添加与阳极和阴极同轴或共面的离子交换膜。膜包围阳极或阴极并在输入料流和电极之间形成半透屏障。cdi和mcdi都将来自溶液的带电盐含量(和其它离子)静电浓缩(通过吸附)到多孔碳电极的具有静电吸引力的表面上。传统cdi池用多孔阴极和阳极,通常含碳材料形成,它们如上所述被一定体积的输入料流隔开。在mcdi中,将互补的阴离子吸引和阳离子吸引膜分别粘贴到阳极和阴极上;这些膜形成各电极与溶液空间之间的屏障。净效应是由于各离子选择性膜提供的增强的吸附选择性,电吸附容量提高。通过各膜的(i)限制从碳电极到输入料流的共离子转移和(ii)用来自输入料流的附加抗衡离子(通过它们穿过该离子选择性膜)平衡从碳表面排出的共离子的能力实现这种提高。mcdi技术中的相关技术包括(i)流电极mcdi(转让给koreainstituteofenergyresearch的eppat2857442),(ii)电位反转以使池再生(转让给voltea的uspat8685255),和(iii)制备阴离子交换膜以减轻共离子排斥(转让给voltea的eppat2641654)。
mcdi以及cdi池传统上对阴极和阳极都使用相同的原始电极材料装配;电容去离子池中的电极必须高度导电和足够多孔以吸附大量离子。在与已知cdi和mcdi技术相比的改进中,本文中对mcdi池公开的发明显示(1)具有当暴露在水溶液下时能够水解以变成带电表面基团的定向表面官能团的电极,和(2)利用这些带电表面基团有效吸引抗衡离子。本发明人已经发现,尽管给定膜的离子选择性,但带电表面基团改变零电荷电位(epzc)的位置(如图9中所示),并且仍可通过epzc影响mcdi性能,因为该溶液与电极直接接触。上文公开的“epzc位移”法也适用于通过确定epzcs的位置和在mcdi运行的充电和放电循环过程中将位移的epzcs与该膜的功能协同组合而改进mcdi性能。这些改进的mcdi池被称作不对称膜电容去离子(“amcdi”)池,相关的“epzc位移”被称作amcdi法。
图26a和26b比较用碳干凝胶(cx)电极形成的传统cdi和mcdi池经使用小时数的性能。cx电极具有标称表面积为~200平方米/克的介孔结构。该电极标作原始,因为它们在此处所示的实验之前没有经过任何处理。mcdi明显胜过cdi,并表现出电导率的更大降低。图27a表明尽管cdi最初在欧姆区中传送更多电子电荷,mcdi在电容区中超过cdi并也可带来如较低最终电流值所示的降低的电荷泄漏。最终,当cdi和mcdi池连续循环时,在cdi池中传递更多电荷,尽管由于较高漏泄电流而在较低效率下。经试验期(图27b),mcdi表现出比cdi好的性能保持(类似于图4中显示的结果)。使用阻抗谱法的运行后epzc分析显示cdi阳极的显著epzc重新定位,而cdi阴极、mcdi阳极和mcdi阴极如图28a–28b和29a–29b中所示表现出部分重新定位(fractionalrelocation)。这意味着mcdi中的膜能够经累积池循环保持epzc位置。
在epzc位置对去离子的影响的进一步验证中,原始和氧化cx电极配对以形成cdi和mcdi池。如通过电化学阻抗谱(eis)确定的原始和氧化电极的epzcs分别为~-0.1v和+0.5vvs.sce电极,意味着在不存在外加电子电荷的情况下和在短路条件下原始电极天然吸附阴离子,而氧化电极天然吸附阳离子。图30a和30b显示,不同于在阳极和阴极位置使用类似原始电极的传统配置,当取而代之在阳极处用原始电极和在阴极处用氧化电极装配cdi和mcdi池由此使工作电压窗口内的抗衡离子过量的电位最大化时,与它们各自的原始-原始配置相比吸附提高。相反,用原始阴极和氧化阳极装配cdi或mcdi池导致降低或倒转的性能。mcdi池配置的电流廓线(图31a和31b)依序显示传递的电子电荷的提高:原始阳极-氧化阴极、原始阳极-原始阴极和氧化阳极-原始阴极。原始阳极-氧化阴极mcdi配置在下文称作不对称mcdi(amcdi),而其cdi对应物是不对称cdi(acdi)。概括mcdi结果的条形图显示在图32a-32c中,并且表明当在阳极处用原始电极和在阴极处用氧化电极装配amcdi池时,与各自的仅原始mcdi配置相比,盐吸附容量提高多达75%。与原始-原始cdi池(~2.5mg/g)相比,盐吸附容量的这种提高可多达200%。
在图30a–30b、图31a–31b和32a–32c中,当用原始阳极和氧化阴极装配池时,工作电压窗口内的抗衡离子过量最大化,并且这一电极配置被认为适用于在1.2v下充电和在0v下放电的运行模式。相反,当用氧化阳极和原始阴极装配池时,工作电压窗口内的共离子过量最大化。这一配置在运行上不适用于电容去离子。在图30a中,对氧化阳极和原始阴极cdi配置观察到倒转廓线,以使放电时的电导率大于充电过程中。但是,安置膜(图30b,mcdi配置)可抑制用氧化阳极和原始阴极的cdi配置观察到的倒转廓线。amcdi的这一实例(原始阳极-氧化阴极mcdi配置)表明在适当配置时,在cdi和mcdi运行中移动epzcs提供显著的附加益处,例如显著改进的脱盐性能。
在具有
为了验证用于改进mcdi性能的epzc位移法不是电极特异性的,将该amcdi方法扩展至
在具有spectracarb碳电极的mcdi中的epzc效应
为了进一步改进amcdi的性能,其用高孔隙率、高表面积(1600平方米/克)spectracarb(sc)电极测试。sc具有微孔结构(图36)且原始sc是供货态的sc。氧化sc经由硝酸处理形成,且它们各自的epzcs为(-)0.1v和(+)0.3vvs.sce参比电极(图36a和36b)。比较两种池组合:原始阳极-原始阴极,和原始阳极-氧化阴极。原始阳极-氧化阴极mcdi(即amcdi)表现出比原始阳极-原始阴极高的电导率降低(图37a)。其标称电吸附容量为~20mg/g(图37b)。amcdi池也传送更多电子电荷,但这两种池都表现出接近1的电荷效率和经试验期的优异稳定性(图37c和37d)和epzc重新定位的减轻。与cdi相比,如果mcdi或amcdi池具有180天的分离寿命(与之前显示的传统cdi的10天相比)并且该单元的初始和更换成本为$10000(由于包含离子交换膜,高于cdi或i-cdi),这相当于$40,556的2年成本,因为该单元需要每180天更换;尽管比i-cdi昂贵,但amcdi与具有$365,000的估算2年成本的传统cdi相比仍是明显的改进。这一估算没有考虑仍是关键值的盐吸附容量,或盐吸附速率。
在单膜运行中改变epzc位置
mcdi和amcdi的一个主要缺点是在形成分离池时需要膜对。但是,使用单膜amcdi(即池中的一个极性的电极被膜覆盖,且该池中的另一极性的电极未被膜覆盖)也有可能实现适当的电极epzc位置以(i)促进孔隙空间中的特定离子过量吸附和(ii)减轻离子排斥。epzc的这种定位足以在装置制造中节省成本的同时增进和保持mcdi性能。用原始sc阳极和阴极构造四种池配置,包括cdi、mcdi、仅cmx的cdi(cmx-cdi)和仅amx的cdi(amx-cdi)。原始sc(epzc=-0.1v)电极在短路电位(eo)下提供过量阴离子,当该电极用作阴极以在1.2v下吸附阳离子时,这也是一种限制。对于单膜cdi池,在负电极处使用该cmx膜,并在正电极处使用amx膜。图38a表明cmx-cdi能够提供与完整mcdi池类似的电导率降低(即提高的离子吸附)。但是,amx-cdi配置没有产生益处,但受困于进一步的性能损失,这可能由于在膜-电极界面处的额外电阻。mcdi更早表现出防止或减轻由电化学反应造成的epzc重新定位。通过阴极处的溶解氧还原平衡碳阳极的电氧化。该膜使阴极缺氧,由此相应地限制阳极氧化。使用原位探针监测池运行过程中的氧响应,并如预期,当阴极被cmx膜覆盖时,在溶解氧中观察到极小扰动,并且考虑到epzcs的类似性,性能与mcdi性能几乎相同(图38a和b)。相反,对于amx-cdi池,性能更差。此外,如图38c中所示,amx-cdi池表现出ph对测得电导率的最大影响;高或低ph暗示羟基或水合氢离子对溶液电导率的较大贡献。使用氧化sc阴极消除阴极限制极大改进amx-cdi池的性能。具有不对称电极的这一配置被标作amx-acdi(图39a-39d)。但是,与传统mcdi池相比仍观察到较大的溶解氧和ph扰动。在图40a-40c中比较cdi、mcdi、amcdi、cmx-cdi、amx-cdi和amx-acdi池的长期性能并可概括如下:amcdi法具有最高容量和效率,但amx-acdi可实现类似性能,尽管在较低效率下(归因于如由溶解氧廓线观察到的对寄生电化学反应的减轻更有限)。cmx-cdi池能够提供与标准mcdi池类似的性能和效率,而amx-cdi表现出所有组合中的最差性能。
不同于在电容去离子池中使用多孔碳的任何之前的研究,本公开的amcdi法(1)利用电极处理制造具有不同epzcs的电极,(2)描述了带来显著性能增强的电极epzc位置和电极配置,(3)描述了带来改进的去离子性能和盐吸附容量的协同电极epzc-膜配置,和(4)证实利用epzc和单膜池组合去离子。该性能增进程序也并非专用于给定类型的原始碳电极并可适用于各种制造商和结构类型(例如圆柱形、织物、平面等)的如本文中公开的电极。这些cdi和mcdi池可用于从任何类型的输入料流,如发电厂废水、用于饮用水净化和软化的储备进料、用于饮用水净化和软化的海水进料、洗衣废水、用于实验室水净化的进料中除去盐和其它离子含量,并可扩展至需要将含盐和/或其它离子的水去离子、净化和/或软化的其它用途。本文中公开的发明具有宽泛的商业意义。此外,用本文中公开的epzc定位方法实现的改进的电吸附容量和电荷效率可显著降低去离子运行过程中的能量消耗和降低总体装置尺寸。
与cdi相比,如果amx-acdi或cmx-acdi池具有180天的分离寿命(与之前显示的传统cdi的10天相比)并且该单元的初始和更换成本为$75,000(由于包含离子交换膜,略高于cdi或i-cdi),这相当于$30,417的2年成本,因为该单元需要每180天更换,与具有$365,000的估算2年成本的传统cdi相比仍是明显的改进。当资本成本较不成问题且装置尺寸和脱盐容量更重要时,使用amx或cmx-acdi代替i-cdi。这一估算没有考虑仍是关键值的盐吸附容量,或盐吸附速率;i-cdi和amx-mcdi/cmx-acdi在这些参数上都胜过cdi和mcdi。曲线图显示在图41中,其描绘随时间的各技术成本,cdi和i-cdi的每单元初始估算值为$5,000,mcdi的每单元初始估算值为$10,000,且amx-mcdi或cmx-mcdi的每单元初始估算值为$7,500。cdi、mcdi、i-cdi和amx-mcdi或cmx-mcdi的预计分离寿命分别为10天、180天、365天和180天。
在cdi、i-cdi、amcdi或其它电容型分离中的能量回收
在本文中公开的所有分离池中,当使用外加电位将池充电时,电荷储存在电极表面,尽管从本体溶液中净分离离子。这意味着在放电过程中,当电位短路或降低(在cdi中解吸离子,在i-cdi中吸附离子)时,可以以电流形式回收能量。尽管在较稀的盐溶液中仍有大的电阻损耗,能量回收仍然相当显著。通过优化电极表面化学和电导率,可使盐吸附/解吸过程与能量回收情形匹配(例如将电容器充电,驱动发动机,驱动逆变器,驱动dc/dc变换器,操作泵),由此优化该分离法的能量成本。本文中注释的表面化学可以更优地从溶液中分离盐,无论是在短路条件下(i-cdi)还是在外加电位下(amcdi、amx-acdi和cmx-acdi),并可以通过将放电和充电池连接在一起而与能量回收操作合并。dc/dc变换器可用于有效转移这一电能并带来更有效的综合水处理/盐分离法。
用具有不同离子吸附容量的电极调节ph
利用表面电荷增强电极和有意定位电极的epzc可以有效调节水溶液的ph。例如,如果使用两个具有正增强表面电荷(更负的epzc)的电极,在电位下的ph会随外加电位提高,并在池短路时降低。在传统cdi池中,当使用原始碳电极时,存在正表面电荷,意味着天然吸附阴离子(例如氯离子)。因此,阴极会限制使用这种cdi池的吸附/分离,其中在阴极处仅发生有限阳离子吸附,而在阳极处发生显著阴离子吸附。在本体溶液中,由于除去比阳离子多的阴离子,ph会提高以保持溶液电中性。cdi池的ph波动显示在图42中。同样地,如果使用两个具有负增强表面电荷的电极,会促进阳离子吸附,且ph会在电位下降低并在池短路时提高。最后,如果如在i-cdi池中那样从溶液中除去类似量的阴离子和阳离子,将使ph波动最小化。由氧化阳极和原始阴极电极构成的i-cdi池的示例性ph波动也显示在图42中。所示ph波动比cdi池中小得多。
通过调节溶液的ph,可以实现在其它水处理系统中几乎不可能的各种分离。例如,硼是典型难从溶液中除去的化合物,因为其在中性溶液中不带电并且也不水合。由于其不水合,反渗透膜法更难将其除去。但是,如果提高溶液中的ph,硼会离子化并可随后使用膜或电容型方法,如cdi、i-cdi、amcdi等分离。由此,如果使用外加电位和具有正表面电荷的电极或通过阴极处的氧还原提高溶液的ph,我们可以以硼酸根离子形式从溶液中除去硼。在图43中显示通过氧还原在阴极处制造氢氧化物以将硼转化成硼酸根和随后利用带电电极从溶液中分离的一般示意图。
- 还没有人留言评论。精彩留言会获得点赞!